
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 1-K
ANNUAL REPORT
ANNUAL REPORT PURSUANT TO REGULATION A OF THE SECURITIES ACT OF 1933
For the fiscal year ended: April 30, 2018
| ALZAMEND NEURO, INC. | ||
| (Exact name of issuer as specified in its charter) |
| Delaware | 2834 | 81-1822909 | ||
|
State or other jurisdiction of incorporation or organization |
(Primary Standard Industrial Classification Code Number) |
(I.R.S. Employer Identification No.) |
|
3802 Spectrum Boulevard, Suite 112C Tampa, Florida 33612 |
| (Full mailing address of principal executive offices) |
| (844) 722-6333 |
| (Issuer’s telephone number, including area code) |
Common
Stock
(Title of each class of securities issued pursuant to Regulation A)
Corporation Service Company
2711 Centerville Road, Suite 400
Wilmington, Delaware 19808
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
Copy to:
Marc Ross, Esq.
Henry Nisser, Esq.
Sichenzia Ross Ference LLP
1185 Avenue of the Americas, 37th Floor
New York, New York 10036
Telephone: (212) 930-9700
ALZAMEND NEURO, INC.
TABLE OF CONTENTS
| Page | |||
| PART II | |||
| Statements Regarding Forward-Looking Information | 2 | ||
| Item 1. | Business | 2 | |
| Item 2. | Management's Discussion and Analysis of Financial Condition and Results of Operations | 39 | |
| Item 3. | Directors, Executive Officers and Corporate Governance | 46 | |
| Item 4. | Security Ownership of Management and Certain Securityholders | 50 | |
| Item 5. | Interest of Management and Others in Certain Transactions | 51 | |
| Item 6. | Other Information | 52 | |
| Item 7. | Financial Statements | 52 | |
| Item 8. | Exhibits | 53 | |
| Index to Financial Statements of Alzamend Neuro, Inc. | F-1 | ||
| 1 |
Part II.
STATEMENTS REGARDING FORWARD-LOOKING INFORMATION
This Annual Report on Form 1-K contains forward-looking statements. All statements other than statements of historical fact are, or may be deemed to be, forward-looking statements. Such forward-looking statements include statements regarding, among others, (a) our expectations about possible business combinations, (b) our growth strategies, (c) our future financing plans, and (d) our anticipated needs for working capital. Forward-looking statements, which involve assumptions and describe our future plans, strategies, and expectations, are generally identifiable by use of the words “may,” “will,” “should,” “expect,” “anticipate,” “approximate,” “estimate,” “believe,” “intend,” “plan,” “budget,” “could,” “forecast,” “might,” “predict,” “shall” or “project,” or the negative of these words or other variations on these words or comparable terminology. This information may involve known and unknown risks, uncertainties, and other factors that may cause our actual results, performance, or achievements to be materially different from the future results, performance, or achievements expressed or implied by any forward-looking statements. These statements may be found in this Annual Report.
Forward-looking statements are based on our current expectations and assumptions regarding our business, potential target businesses, the economy and other future conditions. Because forward-looking statements relate to the future, by their nature, they are subject to inherent uncertainties, risks, and changes in circumstances that are difficult to predict. Our actual results may differ materially from those contemplated by the forward-looking statements as a result of various factors, including, without limitation, changes in local, regional, national or global political, economic, business, competitive, market (supply and demand) and regulatory conditions and the following:
| · | Our ability to effectively execute our business plan; |
| · | Our ability to manage our expansion, growth and operating expenses; |
| · | Our ability to evaluate and measure our business, prospects and performance metrics; |
| · | Our ability to compete and succeed in a highly competitive and evolving industry; |
| · | Our ability to respond and adapt to changes in technology and customer behavior; and |
| · | Our ability to protect our intellectual property and to develop, maintain and enhance a strong brand. |
We caution you therefore that you should not rely on any of these forward-looking statements as statements of historical fact or as guarantees or assurances of future performance. All forward-looking statements speak only as of the date of this Annual Report. We undertake no obligation to update any forward-looking statements or other information contained herein.
Information regarding market and industry statistics contained in this Annual Report is included based on information available to us that we believe is accurate. It is generally based on academic and other publications that are not produced for purposes of securities offerings or economic analysis. Forecasts and other forward-looking information obtained from these sources are subject to the same qualifications and the additional uncertainties accompanying any estimates of future market size, revenue and market acceptance of products and services. Except as required by U.S. federal securities laws, we have no obligation to update forward-looking information to reflect actual results or changes in assumptions or other factors that could affect those statements.
Item 1. Business.
DESCRIPTION OF BUSINESS
In this Annual Report, unless the context requires otherwise, references to the “Company,” “Alzamend,” “we,” “our company” and “us” refer to Alzamend Neuro, Inc., a Delaware corporation.
Company Overview and Description of Business
The Company
The Company was formed on February 26, 2016 as Alzamend Neuro, Inc. under the laws of the State of Delaware. The Company was formed to acquire and commercialize patented intellectual property and know how to prevent, treat and cure the crippling and deadly Alzheimer’s disease (“Alzheimer’s” or “AD”). The Company has developed a unique approach for combating Alzheimer’s, namely through immunotherapy. Current drugs approved by the FDA for Alzheimer’s only address symptoms and provide no benefit to the impaired immune system in Alzheimer’s.
| 2 |
On May 1, 2016, we obtained a royalty-bearing, exclusive worldwide license from the University of South Florida Research Foundation, Inc. (the “University”), to a mutant-peptide immunotherapy that is designed to be used both as a vaccine and prophylactic against Alzheimer’s. This treatment, known as CAO22W, has transitioned from early stage development to an extensive program of preclinical study and evaluation with an anticipated completion date at the end of June 2019. CAO22W will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it can provide us with any revenue. We plan to file an Investigational New Drug Application (“IND”) with the United States Food and Drug Administration (the “FDA”) with respect to CAO22W in the third quarter of 2019 and prepare to conduct a Phase 1 Clinical Trial at Emory University Alzheimer’s Disease Research Center starting in the latter half of 2019. Upon FDA approval of the IND, we plan to work with Dr. Chuanhai Cao, who is associated with the College of Chemistry, a neuroscientist at the University of South Florida Health Byrd Alzheimer’s Institute and the inventor of what we refer to as “CAO22W,” and Dr. Cao’s medical and biotech research team to launch our Phase 1 Clinical Trial with up to 30 human patients.
On July 2, 2018, we obtained two royalty-bearing, exclusive worldwide licenses from the Licensor to a therapy known as LiProSalTM to mitigate extreme agitation and forestall other deterioration as displayed by patients with up to moderate AD. LiProSalTM is an ionic cocrystal of lithium for the treatment of Alzheimer’s and possibly other neurodegenerative diseases. Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of AD. Unlike traditional medications, which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, it exerts neuroprotective effects, in part, by increasing the brain-derived neurotrophic factor leading to restoration of learning and memory based on the results with some of the animal subjects during the preclinical studies. Another neuroprotective mechanism of lithium is attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Alzamend is continuing to work with two of the inventors of this therapeutic, both at the USF Morsani College of Medicine; Jun Tan, PhD, MD, Professor at the College of Medicine Neurosurgery, Endowed Chair, College of Medicine Psychiatry and Behavioral Neurosciences and the Silver Endowed Chair in Developmental Neurobiology, Department of Psychiatry & Behavioral Medicine and Roland D. Shytle, PhD, Associate Professor, Center of Excellence for Aging & Brain Repair. Dr. Tan and Dr. Shytle with Adam J. Smith, PhD., MSBE, Michael Zaworotko, PhD., and Naga Duggirala, PhD., all formerly with the University of South Florida, have designed, synthesized and characterized this ionic cocrystal of lithium, which Alzamend has named LiProSalTM. LiProSalTM has been shown to exhibit improved pharmacokinetics compared to current FDA-approved lithium-based drugs as well as bioactive in many in vitro models of Alzheimer’s disease. LiProSalTM may prove to be a major improvement over current lithium-based treatments and may also represent a means of treating AD.
Technology
The patented solution that Alzamend has licensed and that it will first move to commercialization is an ionic cocrystal of lithium for the treatment of Alzheimer’s disease and a method of preparation for other pharmaceutical and industrial purposes. Lithium salts have a long history of human consumption beginning in the 1800’s. In psychiatry, they have been used to treat mania and as a prophylactic for depression since the mid-20th century. Today, lithium salts are used as a mood stabilizer for the treatment of bipolar disorder. Although the FDA has approved no medications as safe and effective treatments for suicidality, lithium has proven to be the only drug that consistently reduces suicidality in patients with neuropsychiatric disorders. Despite these effective medicinal uses, current FDA-approved lithium pharmaceutics (lithium carbonate and lithium citrate) are limited by a narrow therapeutic window that requires regular blood monitoring of plasma lithium levels and blood chemistry by a clinician to mitigate adverse events. Because conventional lithium salts (carbonate and citrate) are eliminated relatively quickly, multiple administrations throughout the day are required to safely reach therapeutic plasma concentrations. However, existing lithium drugs such as lithium chloride and lithium carbonate suffer from chronic toxicity, poor physicochemical properties and poor brain bioavailability. Because lithium is so effective at reducing manic episodes in patients with bipolar disorder, it is still used clinically despite its narrow therapeutic index. This has led researchers to begin to look for alternatives to lithium with similar bioactivities.
The inventors from the University of South Florida have developed a new lithium cocrystal composition and method of preparation that allows for lower dosages to achieve therapeutic brain levels of lithium for psychiatric disorders, broadening lithium’s therapeutic index. The compound offers improved physiochemical properties compared to existing forms of lithium, giving it the potential to be developed as an anti-suicidal drug or for use against mood disorders. The formulation method may also be used for commercial/industrial applications such as green chemistry, engineering low density porous materials, pesticides/herbicides, explosives/propellants, and electronic materials.
| 3 |
Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of Alzheimer’s disease. Unlike traditional medications which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, it exerts neuroprotective effects, in part, by increasing a brain-derived neurotrophic factor leading to restoration of learning and memory. Another neuroprotective mechanism of lithium is attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Moreover, recent clinical studies suggest that lithium treatment may reduce dementia development while preserving cognitive function and reducing biomarkers associated with AD.
This in mind, this team of inventors from the University of South Florida have designed, synthesized and characterized a new ionic cocrystal of lithium that Alzamend has named and trademarked as “LiProSal”. LiProSalTM has been shown to exhibit improved pharmacokinetics compared to current FDA-approved lithium drugs as well as bioactive in many in vitro models of AD. LiProSalTM is expected to be a major improvement over current lithium-based treatments and may also constitute a means of treating Alzheimer’s disease. The cocrystal technology inherently increases the therapeutic index of lithium providing a greater bioavailability and biodistribution in comparison to more traditional lithium dosage forms. Lithium has been marketed for over 35 years and human toxicology regarding lithium use has been well characterized. We plan to use the existing data available in the public domain supplanted with the knowledge that FDA has about lithium to preclude a need to conduct any further preclinical studies and to submit a pre-IND application to the FDA in early February. Upon receiving feedback from the FDA from the pre-IND, Alzamend is anticipating beginning the process of finalizing the IND application and receiving approval to begin a Phase 1 Clinical Trial with human subjects within the first half of 2019. In 2017, the FDA issued a new “FastTrack” protocol specific to cocrystal based new drug (an “NDA”) and abbreviated new drug applications (an “ANDA”) which expedites the FDA-approval process and timeline, which will be a key component of our FDA IND application for LiProSalTM.
LiProSalTM will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it and any successors could provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval for and commercialize LiProSalTM, our long-term business plans will not be met, and the Company may be unable to generate the revenue it has forecasted for many years, if at all. We do not anticipate that we will generate our maximum revenue for several years, or that we will achieve profitability for this therapeutic at least a few years after generating material revenue, if at all. If we are unable to generate revenue, we may not be able to pursue any expansion of the Company or acquire additional IP as we have planned, we will not become profitable with this therapeutic, and we may be unable to continue our operations.
The other patented solution that Alzamend has licensed to move to commercialization is a mutant-peptide immunotherapy that is designed to be used both as a vaccine and prophylactic against Alzheimer’s, CAO22W, was developed by Dr. Cao Assistant Professor, USF Health, Assistant Professor, Morsani College of Medicine, Neurology and Associate Professor, Pharmaceutical Sciences, USF College of Pharmacy. This therapy is intended to work by stimulating the body’s own immune system to prevent the formation and breakdown of beta amyloids, which build up in the brain to form a “plaque,” and subsequently block the neurological brain signals, ultimately leading to the symptoms and onset of Alzheimer’s. Immunotherapy is the “treatment of disease by inducing, enhancing, or suppressing an immune response.” Immunotherapies that are designed to elicit or amplify an immune response are classified as activation immunotherapies, whereas immunotherapies that reduce or suppress are classified as suppression immunotherapies. We believe that strategies to strengthen the immune system in the elderly, who are most susceptible to the development of Alzheimer’s, could greatly enhance the effectiveness of immune-based approaches against Alzheimer’s. Our novel immune-based methodology attempts to inhibit the natural process of immunological aging by restoring the balance of immune system through immunomodulation. An exclusive license agreement with sublicensing terms (Agreement #LIC16118) was made effective on May 1, 2016, as amended on August 17, 2017 and May 7, 2018 (the “Effective Date”) by and between the University, which is a direct support organization of the University of South Florida, and the Company.
Beta amyloid protein has been directly linked to Alzheimer’s disease and the associated neurofibrillary tangles formation seen in Alzheimer’s patients. Specifically, increased levels of extracellular plaques in the brain composed of amyloid beta peptide 1-42 are seen in Alzheimer’s patients when compared to healthy people. Attempts have been made to help inhibit plaque formation by reducing the amount of amyloid beta peptide 1-42 through vaccines that generate an immune response to the protein. The challenge has been that though effective in reducing the amount of the protein, the inflammatory response has been such that the intended benefits are not seen. These vaccines have used an adjuvant, or helper, to generate the necessary immune response and it is believed that this adjuvant triggers the unwanted surplus inflammation. We have licensed rights to a vaccine using autologous cells that does not require an adjuvant and therefore, we believe will trigger the immune response, which should help eliminate the amyloid beta peptide 1-42 without generating the excess inflammation and therefore, have a positive clinical effect. We believe that the vaccine, in addition to dealing with plaque formation, also ameliorates the impaired immune system that is thought to be the major issue in Alzheimer’s patients.
| 4 |
Our data have demonstrated that these mutant-peptide sensitized dendritic cells (“DC”) can act as a vaccine to generate a durable antibody response, as well as enhance the number of CD8+ T-cells and increase the lifespan of CD8+ cells (T and DCs cells), compared to control subjects. These studies will provide a further rationale and impetus for using this novel vaccine to determine potential efficacy in human clinical trials against Alzheimer’s.
CAO22W has been researched for more than ten (10) years and is currently in the midst of completing its preclinical development and has started both its pre-IND and IND application process to the FDA which is managed by TAMM Net, Inc. an experienced and highly-regarded regulatory consultancy firm. In November 2018, the Company began a toxicological preclinical 6-month study for CAO22W with Charles River Laboratories, Inc. (“CRL”) in compliance with FDA IND requirements. Upon conclusion of this toxicological study, anticipated to occur in May 2019, we have determined that the Company will be ready to begin the process of finalizing the IND application process and move quickly forward to begin a Phase 1 Clinical Trial with human subjects. CAO22W will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it and any successors could provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval for and commercialize CAO22W, our long-term business plans will not be met, and the Company may be unable to generate the revenue it has forecasted for CAO22W for many years, if at all. We do not anticipate that we will generate our maximum revenue for several years, or that we will achieve profitability for this therapeutic at least a few years after generating material revenue, if at all. If we are unable to generate revenue, we may not be able to pursue any expansion of the Company or acquire additional IP as we have planned, we will not become profitable with this therapeutic, and we may be unable to continue our operations.
Market
Currently, Alzheimer’s is the 6th leading cause of death in the U.S. and when extrapolated globally, the market for preventions, treatments, and cures of this crippling disease is massive. The Alzheimer’s Association estimates that the cost of caring for people with Alzheimer’s and other dementias will reach $277 billion in 2018 and that by 2050, these costs may rise as high as $1.1 trillion. Since 1990, life expectancy has increased by 6 years and the worldwide average continues to increase. With the increase in the mean age of the population in developed countries, the prevalence of deteriorating neurological diseases has also increased. According to the Alzheimer’s Association, in the United States alone, 1 in 10 persons over the age of 65 have Alzheimer’s disease, with more than 5.7 million Americans living with it. It is estimated that this number will increase to more than 14 million by 2050 if a cure is not found. Many Alzheimer’s related associations believe the actual number of adults with AD may be as much as 5 times more or 30 million since current statistics do not take in account deaths from complications or from related diseases like pneumonia or heart attack. These death certificates only list the most immediate cause. The fastest growing age group in the United States is the “over 85” group with 1 in 3 having Alzheimer’s. Women are 2½ times more likely to die from Alzheimer’s than from cancer.
The rate of deaths related to Alzheimer's disease increased by 54.5 percent over 15 years, according to a new report issued on May 27, 2017 from the U.S. Centers for Disease Control and Prevention. There were 93,541 deaths related to Alzheimer's disease in 2014, a rate of 25.4 deaths per 100,000 people, up from 44,536 deaths in 1999, a rate of 16.5 deaths per 100,000 people, according to the report. The disease currently affects an estimated 5.7 million people in the U.S., but that number is expected to rise dramatically in people over the age of 65 to 13.8 million in 2050. The researchers examined death certificate data from the National Vital Statistics System to reach their findings.
Every 65 seconds, someone in the United States develops AD. Of the 10 most fatal diseases in the United States, Alzheimer’s disease is the only one with no cure, no known way of deceleration and no known means of prevention. Alzamend was formed to commercialize patented intellectual property in this space, by funding it from its present state through clinical trials administered by the FDA and ultimately, if successful, potentially to the global market.
Business Plans
Our plan of operations is currently focused on the development of both our therapeutic candidates which are at different stages in development. The Company preparing a Pre-IND application to the FDA for our first product candidate, LiProSalTM, an ionic cocrystal of lithium for the treatment of Alzheimer’s disease and a method of preparation for other pharmaceutical and industrial purposes, anticipating its submission in the month of March 2019.
Our second product candidate, CAO22W, is in the midst of completing a preclinical toxicology study with CRL. Alzamend, working with its regulatory consultant, TAMM Net, has already begun the both the Pre-IND and the IND application process. At this time, the goal of these efforts is to initiate a Phase 1 Clinical Trial expected to have up to 30 human participants, contingent upon guidance from the FDA. The FDA will provide the Company with the final determination on how many participants will be required along with all other details, requirements and parameters that comprise this Phase 1 Clinical Trial.
| 5 |
The Company has engaged Emory University, located in Atlanta, Georgia, to develop and plan the Phase 1 Clinical Trial protocols, processes and plan. Dr. Ihab Hajjar, Neurologist at the Emory Clinic, has been selected to be the Lead Investigator for this set of preclinical activities. The Company has also retained a division of the international Swiss manufacturer, Lonza, to develop its manufacturing protocols, processes and procedures for CAO22W. Lonza is the worldwide leader in producing immunological proprietary and contracted pharma solutions. Alzamend anticipates selecting Emory University as the host for the Phase 1 Clinical Trial and to be led by Dr. Hajjar.
In November 2018, the Company adopted a Charter for its Scientific Advisory Board (“SAB”) and is now completing the process of selecting the initial SAB members. The SAB is an important advisory group whose members will be chosen from among physicians and medical experts with various medical and clinical specializations, including extensive experience with either AD or other neurological diseases. It is from this effort, that Alzamend endeavors to gather and retain the best sources of experience and knowledge, so it may confidently rely on this group of experts to help guide it through all of its scientific and manufacturing endeavors.
The continuation of our current plan of operations to completing our IND application to progress to start the series of human clinical trials for each of our therapeutics, requires us to raise significant additional capital promptly. If we are successful in raising capital, we believe that the Company will have sufficient cash resources to fund its operations for the next twelve months or more.
Because our working capital requirements depend upon numerous factors, including the progress of our preclinical and clinical testing, timing and cost of obtaining regulatory approvals, changes in levels of resources that we devote to the development of manufacturing and marketing capabilities, competitive and technological advances, status of competitors, and our ability to establish collaborative arrangements with other organizations, we will require additional financing to fund future operations.
FDA consulting and active project planning management
The Company has retained TAMM Net, Inc., an experienced GMP, 10-year old consulting firm based in Georgia for project management; to lead, develop and manage our preclinical and clinical efforts, extending from the current status of each product candidate through the exit or commercialization of the technologies licensed by the Company. Alzamend may retain an experienced Canadian Health and European Union consulting firm to commercialize these same technologies for these geographic markets.
Funding new AD research and acquisition of licenses to treat or cure AD
Alzamend has committed to funding new research projects from Dr. Cao and his medical team for up to the next three years or more.
Alzamend obtained two royalty-bearing, exclusive worldwide licenses from the Licensor, originally called LisPro, now given the trade name of LiProSalTM, a cocrystal biologic therapy to mitigate extreme agitation and forestall further deterioration of memory as displayed by patients with up to moderate AD effective as of July 2, 2018.
Alzamend is dedicated to acquiring and supporting new research to treat or cure AD and reserves the right to evaluate and pursue each opportunity as it may arise.
Establishment of advisory board, initial meetings, corporate development and initial consulting
The Company intends to recruit top notch leaders in the Alzheimer’s and business communities, who will bring their knowledge and experience to assist the Company in developing a means for the prevention, treatment and cure for Alzheimer’s, to serve either on the Company’s SAB or its Board of Directors (the “Board”). The Company established its SAB in November 2018 and is in the process of retaining its first initial members to its SAB.
| 6 |
Intellectual Property and Licensing Agreements
Licensing Fees and ongoing project support for University of South Florida and the USF Health Byrd Alzheimer’s Institute
There are certain license fees and milestone payments required to be paid for the licensing of the CAO22W technology, pursuant to the terms of the Standard Exclusive License Agreement with Sublicensing Terms dated 05/01/2016, License #16118, (the “CAO22W License Agreement”) with the Licensor and the University. In addition to royalty payments of 4% on net sales of products developed from the licensed technology, the Company was required to pay a license fee of $100,000 on June 25, 2016 and December 31, 2016 totaling $200,000. As an additional licensing fee, the Licensor received 3,601,809 shares of the Company’s Common Stock equal to five percent (5%) of the sum of the total number of issued and outstanding shares plus any securities that are convertible into or exercisable or exchangeable for shares of common stock, subject to adjustment for additional issuances until such time as the Company has received a total of $5 million in cash in exchange for the Company’s equity securities. Additionally, the Company is required to pay milestone payments on the due dates to Licensor for the license of the technology, as follows:
| Payment | Due Date | Event | ||||
| $ | 50,000 | September 30, 2019 | Upon IDN application filing | |||
| $ | 50,000 | 12 months from IND application filing date | Upon first dosing of patient in first Phase I Clinical Trial | |||
| $ | 175,000 | 12 months from first patient dosed in Phase I | Upon Completion of first Phase I Clinical Trial | |||
| $ | 500,000 | 24 months from completion of first Phase I Trial | Upon Completion of first Phase II Clinical Trial | |||
| $ | 1,000,000 | 12 months from completion of the first Phase II Clinical Trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA BLA Approval | |||
None of these milestones was met as of the date of this Annual Report. If we fail to meet a milestone by its specified date, the Licensor may terminate the CAO22W License Agreement.
Licensor was also granted a preemptive right to acquire such shares of Common Stock or other equity securities that may be issued from time to time by the Company while Licensor remains the owner of any equity securities of the Company. Further, if the Company issues equity securities at a price per share that is less than the price paid by purchasers in a transaction for aggregate consideration of at least $5,000,000 (the “Investment Price”), then the number of shares of Common Stock owned by Licensee shall be increased upon such issuance. The amount of the increase shall be determined by multiplying the number of shares then owned by Licensor by a fraction; the numerator of which shall be equal to the number of shares of Common Stock outstanding immediately after the issuance of additional shares of Common Stock, and the denominator of which shall be equal to the sum of (i) the number of shares of Common Stock outstanding immediately prior to the issuance of additional shares of Common Stock plus (ii) the number of shares of Common Stock which the aggregate consideration for the total number of additional shares of Common Stock so issued would purchase at the Investment Price.
Additionally, Alzamend is striving to support the ongoing work performed at the USF Health Byrd Alzheimer’s Institute, a multi-disciplinary center at the University of South Florida, required for the commercialization of the CAO22W technology and further research associated with other technologies that Alzamend has and will have a first right of refusal to commercialize. Alzamend has committed to fund new research projects with Dr. Cao and his medical team over the next three years or more as a part of these efforts. In addition, Alzamend has submitted an application for $150,000 in funds from The Florida High Tech Corridor Matching Grants Research Program. The grant is for one year and can be renewed annually with Alzamend working with the USF Senior Sponsored Research Administrator and the CRA-USF Matching Grants Program Director for the Florida High Tech Corridor to secure this grant.
There are certain license fees and milestone payments required to be paid for the licensing of the LiProSalTM technology, pursuant to the terms of the Standard Exclusive License Agreements with Sublicensing Terms, both dated July 2, 2018 (the “LiProSalTM License Agreements”) with the Licensor and the University. In addition, a royalty payment of 3% is required pursuant to License #18110 while License #1811 requires a royalty payment of 1.5% on net sales of products developed from the licensed technology. For the two LiProSalTM based licenses, in the aggregate, the Company is required to pay initial license fees of $50,000 no later than July 31, 2018 and $150,000 no later than October 31, 2018, although the Company is in discussions with the University to defer the payment due date of the initial license fees. As an additional licensing fee, the Licensor is entitled to receive that number of shares of Common Stock equal to three percent (3%) of the sum of the total number of such issued and outstanding shares. Additionally, the Company is required to pay milestone payments on the due dates to Licensor for the license of the technology, as follows:
| 7 |
| Payment | Due Date | Event | ||||
| $ | 50,000 | November 1, 2019 | Pre-IND meeting | |||
| $ | 65,000 | 6 months from IND filing date | IND application filing | |||
| $ | 190,000 | 12 months from IND filing date | Upon first dosing of patient in a clinical trial | |||
| $ | 500,000 | 12 months from first patient dosing | Upon Completion of first clinical trial | |||
| $ | 1,250,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 8 years from the Effective Date of the Agreement | Upon FDA Approval | |||
Our Science
Market Opportunity
The Alzheimer’s Association estimates that the cost of caring for people with Alzheimer’s will reach $277 billion dollars in 2018 and by 2050, these costs may rise as high as $1.1 trillion. Currently, Alzheimer’s is the 6th leading cause of death in the U.S. and when extrapolated globally, the market for preventions, treatments, and cures of this crippling disease is massive. Alzamend was formed to commercialize patented intellectual property in this space, by funding it from its present state through FDA Clinical Trials and ultimately, if successful, to the global market. Additionally, Alzamend is supporting ongoing research at the USF Health College of Medicine, and plans to support others with first rights of refusal on technologies for treating terminal diseases.
In an article jointly issued on April 8, 2016, Allergan and Heptares cited currently significant unmet medical needs and a heavy economic burden caused by cognitive impairment and dementia across multiple diseases, noting that currently available drugs for treating Alzheimer’s disease provide limited and transient effects on cognition. They cite projections of healthcare costs, including nursing home care, associated with Alzheimer’s and dementia (currently estimated to be in excess of $640 billion for North America, Western Europe, and Asia-Pacific), that are continuing to grow based on data from the World Health Organization, Alzheimer’s Disease International, the National Institute of Mental Health, and the Lewy Body Dementia Association.
| 8 |
This medical shortfall puts a spotlight on an urgent need for development of new therapies capable of treating the estimated more than 45 million people worldwide suffering from dementia today - 5.7 million in North America, 7.5 million in Western Europe, and 3.6 million in Asia-Pacific - a number expected to increase to more than 130 million by 2050. Alzheimer’s is the most common cause of dementia, estimated to be associated with some 60 to 70 percent of cases. An additional estimated 1.4 million patients in the U.S. suffer from Lewy body dementia. The potential marketplace for a commercialized therapy or treatment would be tremendously significant with large financial support available from numerous national and international pharmaceutical companies and various governments and worldwide agencies.
These statistics were recently affirmed domestically in an article regarding the death rate and pervasiveness of Alzheimer’s. The rate of deaths related to Alzheimer's disease jumped by 54.5 percent over 15 years, according to a new report issued on May 27, 2017 from the U.S. Centers for Disease Control and Prevention. There were 93,541 deaths related to Alzheimer's disease in 2014, a rate of 25.4 deaths per 100,000 people, up from 44,536 deaths in 1999, a rate of 16.5 deaths per 100,000 people, according to the report. The disease currently affects an estimated 5.3 million people in the U.S., but that number is expected to rise dramatically in people over the age of 65 to 13.8 million in 2050. The researchers examined death certificate data from the National Vital Statistics System to reach their findings.
Manufacturing
We do not have any in-house manufacturing capabilities. The Company intends to outsource the manufacturing of its products to third party contractors, with special capabilities to manufacture chemical drugs and biologic drug candidates for submission and clinical testing under FDA guidelines. There are several sources of manufacturing available once a therapy or treatment can achieve Phase 2 study as identified in a publication by Pharma.org released in 2013 (http://www.phrma.org/sites/default/files/Alzheimer's%202013.pdf).
For CAO22W, the Company has selected the worldwide leader and authority in the manufacturing of immunological peptides, Lonza, which is a Swiss multinational, chemicals and biotechnology company, headquartered in Basel, with major facilities in Europe, North America and South Asia. Lonza was established under that name in the late 19th-century in Switzerland. The company provides product development services to the pharmaceutical and biologic industries, including organic, fine and performance chemicals, custom manufacturing of biopharmaceuticals, chemical synthesis capabilities, detection systems and services for the bioscience sector.
Distribution & Marketing
We intend to develop CAO22W and LiProSalTM through successive de-risking milestones towards regulatory approval and seek marketing approval of CAO22W and LiProSalTM or effect partnering transactions with biopharmaceutical companies seeking to strategically fortify pipelines and fund the costly later-stage clinical development required to achieve successful commercialization. We do not anticipate selling products directly into the marketplace, although we may do so depending on market conditions. Our focus is to strategically effect partnering transactions which will provide distribution and marketing capabilities to sell products into the marketplace.
Government Regulation
Clinical trials, the pharmaceutical approval process, and the marketing of pharmaceutical products, are intensively regulated in the U.S. and in all major foreign countries.
Human Health Product Regulation in the U.S.
In the U.S., the FDA regulates pharmaceuticals under the Federal Food, Drug, and Cosmetic Act and related regulations. Pharmaceuticals are also subject to other federal, state, and local statutes and regulations. Failure to comply with applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the imposition by the FDA of an Institutional Review Board (“IRB”), a clinical hold on trials, a refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The FDA and comparable regulatory agencies in state and local jurisdictions impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising and promotion of our products.
| 9 |
The FDA’s policies may change, and additional government regulations may be enacted that could prevent or delay regulatory approval of new disease indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or elsewhere.
Marketing Approval
The process required by the FDA before human health care pharmaceuticals may be marketed in the U.S. generally involves the following:
| · |
nonclinical laboratory and, at times, animal tests;
| |
| · |
adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use or uses;
| |
| · |
pre-approval inspection of manufacturing facilities and clinical trial sites; and
| |
| · | FDA approval of a Biologics License Application (“BLA”), which must occur before a drug can be marketed or sold. |
We will need to successfully complete extensive clinical trials in order to be in a position to submit a BLA or NDA to the FDA. We must reach agreement with the FDA on the proposed protocols for our future clinical trials in the U.S. A separate submission to the FDA must be made for each successive clinical trial to be conducted during product development. Further, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that site, and an informed consent must also be obtained from each study subject. Regulatory authorities, a data safety monitoring board or the sponsor, may suspend or terminate a clinical trial at any time on numerous grounds.
For purposes of BLA or NDA approval for human health products, human clinical trials are typically conducted in phases that may overlap.
| · |
Phase I. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients.
| |
| · |
Phase II. This phase involves trials in a limited subject population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. Phase II studies may be sub-categorized to Phase IIa studies which are smaller, pilot studies to evaluate limited drug exposure and efficacy signals, and Phase IIb studies which are larger studies testing more rigorously both safety and efficacy.
| |
| · | Phase III. This phase involves trials undertaken to further evaluate dosage, clinical efficacy and safety in an expanded subject population, often at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
All of these trials must be conducted in accordance with Good Clinical Practice (“GCP”) requirements in order for the data to be considered reliable for regulatory purposes.
New Drug and Biologics License Applications
In order to obtain approval to market a pharmaceutical in the U.S., a marketing application must be submitted to the FDA that provides data establishing to the FDA’s satisfaction the safety and effectiveness of the investigational drug for the proposed indication. Each NDA or BLA submission requires a substantial user fee payment unless a waiver or exemption applies (such as with the Orphan Drug Designation discussed below). The NDA or BLA submission fee currently exceeds $1,958,000, and the manufacturer and/or sponsor under an approved NDA or BLA are also subject to annual product and establishment user fees, currently exceeding $98,000 per product and $526,000 per establishment. These fees are typically increased annually. The NDA or BLA includes all relevant data available from pertinent non-clinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators.
| 10 |
The FDA will initially review the NDA or BLA for completeness before it accepts it for filing. The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that the application is sufficiently complete to permit substantive review. After the NDA or BLA submission is accepted for filing, the FDA reviews the NDA or BLA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with current Good Manufacturing Practices (“cGMP”) to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Based on pivotal Phase III trial results submitted in an NDA or BLA, upon the request of an applicant, the FDA may grant a Priority Review designation to a product, which sets the target date for FDA action on the application at six to eight months, rather than the standard ten to twelve months. The FDA can extend these reviews by three months. Priority Review is given where preliminary estimates indicate that a product, if approved, has the potential to provide a significant improvement compared to marketed products or offers a therapy where no satisfactory alternative therapy exists. Priority Review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
After the FDA completes its initial review of an NDA or BLA, it will communicate to the sponsor that the drug will either be approved, or it will issue a complete response letter to communicate that the NDA or BLA will not be approved in its current form and inform the sponsor of changes that must be made or additional clinical, nonclinical or manufacturing data that must be received before the application can be approved, with no implication regarding the ultimate approvability of the application.
Before approving an DNA or BLA, the FDA will inspect the facilities at which the product is manufactured, even if such facilities are located overseas. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications.
Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical sites to assure compliance with GCP. If the FDA determines that any of the application, manufacturing process or manufacturing facilities is not acceptable, it typically will outline the deficiencies and often will request additional testing or information. This may significantly delay further review of the application. If the FDA finds that a clinical site did not conduct the clinical trial in accordance with GCP, the FDA may determine that if the data generated by the clinical site should be excluded from the primary efficacy analyses provided in the NDA or BLA. Additionally, notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process for a drug requires substantial time, effort and financial resources, and this process may take up to several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products.
The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called Phase IV studies may be made a condition to be satisfied for continuing drug approval. The results of Phase IV studies can confirm the effectiveness of a product candidate and can provide important safety information. In addition, the FDA now has express statutory authority to require sponsors to conduct post-market studies to specifically address safety issues identified by the agency. Any approvals that we may ultimately receive could be withdrawn if required post-marketing trials or analyses do not meet the FDA requirements, which could materially harm the commercial prospects for LiProSalTM or CAO22W.
| 11 |
The FDA also has authority to require a Risk Evaluation and Mitigation Strategy (“REMS”) from manufacturers to ensure that the benefits of a drug or biological product outweigh its risks. A sponsor may also voluntarily propose a REMS as part of the NDA or BLA submission. The need for a REMS is determined as part of the review of the NDA or BLA. Based on statutory standards, elements of a REMS may include “dear doctor letters,” a medication guide, more elaborate targeted educational programs, and in some cases restrictions on distribution. These elements are negotiated as part of the NDA or BLA approval, and in some cases if consensus is not obtained until after the Prescription Drug User Fee Act review cycle, the approval date may be delayed. Once adopted, REMS are subject to periodic assessment and modification.
Even if CAO22W or LiProSalTM receive regulatory approval, the approval may be limited to specific disease states, patient populations and dosages, or might contain significant limitations on use in the form of warnings, precautions or contraindications, or in the form of onerous risk management plans, restrictions on distribution, or post-marketing study requirements. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delay in obtaining, or failure to obtain, regulatory approval for CAO22W or LiProSalTM, or obtaining approval but for significantly limited use, would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the U.S. National Institutes of Health. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed until the product or new indication being studied has been approved. Competitors may use this publicly-available information to gain knowledge regarding the design and progress of our development programs.
The Drug Price Competition and Patent Term Restoration Act
The Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act, requires pharmaceutical companies to divulge certain information regarding their products which has the effect of making it easier for other companies to manufacture generic drugs to compete with those products.
Patent Term Extension. After an NDA or BLA approval, owners of relevant drug patents may apply for up to a five-year patent extension. The allowable patent term extension is calculated as half of the drug’s testing phase - the time between IND submission and NDA or BLA submission - and all of the review phase - the time between either NDA or BLA submission and approval up to a maximum of five years. The time can be shortened if FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the PTO must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which a NDA or BLA has not been submitted.
Environmental Regulations. The U.S. generally requires an environmental assessment, which discusses a company’s proposed action, possible alternatives to the action, and whether the further analysis of an environmental impact statement is necessary. Certain exemptions are available from the requirement to perform an environmental assessment and an environmental impact statement. Once an exemption is claimed, a company must state to the FDA that no extraordinary circumstances exist that may significantly affect the environment. We may claim an exemption, under the category for biologic products, from the requirement to provide an environmental assessment and an environmental impact statement for CAO22W or LiProSalTM and may furthermore state to the FDA that to our knowledge, no extraordinary circumstances exist that may significantly affect the environment.
| 12 |
FDA Post-Approval Requirements
Following the approval of an NDA or BLA, the FDA continues to require adverse event reporting and submission of periodic reports. The FDA also may require post-marketing testing, known as Phase IV testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMP. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered.
Patient Protection and Affordable Care Act
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “ACA”), which includes measures that have or will significantly change the way health care is financed by both governmental and private insurers, became law in the U.S. The ACA is a sweeping measure intended to expand health care coverage within the U.S., primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. The ACA has significantly impacted the pharmaceutical industry. The ACA will require discounts under the Medicare drug benefit program and increased rebates on drugs covered by Medicaid. In addition, the ACA imposes an annual fee, which will increase annually, on sales by branded pharmaceutical manufacturers. At this time, the financial impact of these discounts, increased rebates and fees and the other provisions of the ACA on our business are unclear. However, the fees, discounts and other provisions of this law are expected to have a significant negative effect on the profitability of pharmaceuticals.
Human Health Product Regulation in the European Union
In addition to regulations in the U.S., we may eventually be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the U.S. have a process that requires the submission of a clinical trial application prior to the commencement of human clinical trials. In Europe, for example, a Clinical Trial Application (“CTA”) must be submitted to the competent national health authority and to independent ethics committees in each country in which a company intends to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed in that country.
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the EU Member States resulting from the national implementation of underlying EU legislation. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of an investigational drug under EU regulatory systems, we must submit a marketing authorization application. This application is similar to the BLA in the U.S., with the exception of, among other things, country-specific document requirements. Drugs can be authorized in the EU by using (i) the centralized authorization procedure, (ii) the mutual recognition procedure, (iii) the decentralized procedure or (iv) national authorization procedures.
The European Medicines Agency (“EMA”) implemented the centralized procedure for the approval of human drugs to facilitate marketing authorizations that are valid throughout the EU. This procedure results in a single marketing authorization granted by the European Commission that is valid across the EU, as well as in Iceland, Liechtenstein and Norway (the “European Community”). The centralized procedure is compulsory for human drugs that are: (i) derived from biotechnology processes, such as genetic engineering, (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases, (iii) officially designated orphan drugs, and (iv) advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may at the request of the applicant also be used for human drugs which do not fall within the above mentioned categories if the human drug (a) contains a new active substance which, on the date of entry into force of Regulation (EC) No. 726/2004, was not authorized in the European Community; or (b) the applicant shows that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization in the centralized procedure is in the interests of patients at European Community level.
| 13 |
Under the centralized procedure in the EU, the maximum timeframe for the evaluation of a Marketing Authorization Application (“MAA”) by the EMA is 210 days, though the date count stops whenever the Committee for Medicinal Products for Human Use (“CHMP”) asks the applicant for additional written or oral information, with adoption of the actual marketing authorization by the European Commission thereafter. Accelerated evaluation might be granted by the CHMP in exceptional cases, as when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: (i) the seriousness of the disease to be treated; (ii) the absence of an appropriate alternative therapeutic approach; and (iii) anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter. We plan to submit an application for marketing authorization in the United States for LiProSalTM in the first half of 2019 and submit an application for marketing authorization in the United States for CAO22W shortly thereafter in the second half of 2019.
The Mutual Recognition Procedure (“MRP”), for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the EU. Basically, the MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products and is based on the principle of recognition of an already existing national marketing authorization by one or more Member States.
The characteristic of the MRP is that the procedure builds on an already existing marketing authorization in a Member State of the EU that is used as reference in order to obtain marketing authorizations in other EU Member States. In the MRP, a marketing authorization for a drug already exists in one or more Member States of the EU and subsequently marketing authorization applications are made in other EU Member States by referring to the initial marketing authorization. The Member State in which the marketing authorization was first granted will then act as the reference Member State. The Member States where the marketing authorization is subsequently applied for act as concerned Member States.
The MRP is based on the principle of the mutual recognition by EU Member States of their respective national marketing authorizations. Based on a marketing authorization in the reference Member State, the applicant may apply for marketing authorizations in other Member States. In such case, the reference Member State shall update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all Member States, together with the approved summary of product characteristics, labeling and package leaflet. The concerned Member States then have 90 days to recognize the decision of the reference Member State and the summary of product characteristics, labeling and package leaflet. National marketing authorizations shall be granted within 30 days after acknowledgement of the agreement.
Should any Member State refuse to recognize the marketing authorization by the reference Member State, on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within a timeframe of 60 days, Member States shall, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products.
Human Health Product Regulation in the Rest of World
For other countries outside of the EU, such as countries in Eastern Europe or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, again, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Other Regulatory Considerations
Labeling, Marketing and Promotion
Once an NDA or BLA is approved, a product will be subject to certain post-approval requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of pharmaceuticals, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities on the internet and elsewhere.
| 14 |
While doctors are free to prescribe any pharmaceutical approved by the FDA for any use, a company can only make claims relating to the safety and efficacy of a pharmaceutical that are consistent with the FDA approval, and is only allowed to actively market a pharmaceutical for the particular indication approved by the FDA. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA/BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing supplements as it does in reviewing NDAs.
In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties. Government regulators recently have increased their scrutiny of the promotion and marketing of pharmaceuticals.
Anti-Kickback and False Claims Laws
In the U.S., we are subject to complex laws and regulations pertaining to health care “fraud and abuse,” including, but not limited to, The Medicare and Medicaid Patient Protection Act of 1987 (otherwise known as the federal “Anti-Kickback Statute”), the federal False Claims Act, state false claims acts and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular pharmaceutical, for which payment may be made under a federal health care program, such as Medicare or Medicaid.
The federal False Claims Act prohibits anyone from knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including pharmaceuticals, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, beginning in 2013, a similar federal law requires manufacturers to track and report to the federal government certain payments made to physicians and teaching hospitals made in the previous calendar year. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state, and soon federal, authorities.
Other Health Care Laws and Compliance Requirements
In the U.S., our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the U.S. Department of Health and Human Services (e.g., the Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992 (“VHCA”), each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements will apply. Under the VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including U.S. Department of Veteran Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Recent legislative changes require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
| 15 |
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors that ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives. Other legislation has been enacted in certain states prohibiting pharmacies and other health care entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection and unfair competition laws.
Diagnostics for Alzheimer’s Disease
Alzheimer’s is a chronic neurodegenerative disorder affecting millions of people worldwide. It is the number one form of dementia in the world. The risk of being afflicted with Alzheimer’s increases with age, with one in ten people over the age of 65 having the disease. The prevalence of the disease is approximately 5.7 million individuals in the US. Alzheimer’s is also the sixth leading cause of death across all ages in the United States [AA2013: 113], and its prevalence is expected to quadruple by 2050. On the other hand, the incidence (or rate at which new cases of disease develop) is age dependent with approximately 53 new cases per 1,000 people age 65 to 74, 170 new cases per 1,000 people age 75 to 84, and 231 new cases per 1,000 people age 85 and older, with 454,000 new cases occurring in 2010 [Alzheimer’s Association, 2013 Alzheimer’s Disease Facts and Figures, Alzheimer’s & Dementia, Volume 9, Issue 2]. It is estimated that the cost of caring for people with Alzheimer’s and other dementias will increase from an estimated $277 billion in 2018 to a projected $1.1 trillion per year by 2050 with Medicare and Medicaid covering approximately 70% of such costs. Over 16 million Americans provide unpaid care for people with Alzheimer’s disease or other dementias. It is estimated that in 2017 caregivers to people with Alzheimer’s provided 18.4 billion hours of care valued at $232 billion.
The cause and progression of Alzheimer's disease are not well understood. As of 2015, more than 1,200 clinical trials have been or are being conducted to find ways to treat the disease, but it is unknown if any of the tested treatments will work.
According to the Alzheimer’s Foundation of America, it is widely accepted that with the increasing trend towards a longer lifespan coupled with the baby-boomer population approaching retirement, the incidence of Alzheimer’s is likely to double in the next 20 years. The exponential increase in the expected number of patients presenting with Alzheimer’s not only represents a major area of unmet medical need, but it also represents a significant market opportunity for diagnostics for this disease. Alzheimer’s biomarker sales in 2011 were reported at $1.5 billion but are expected have doubled in 2018 to over $3 billion. (BCC research 2013, Advances in biomarker and monitoring diagnostics: Great markets, not so great health effects by Bjørn Hofmann PhD and H. Gilbert Welch MD, MPH, 2017).
Current clinical research focuses on the early phases of the disease. However, no accurate and convenient tools are available today for pre-dementia diagnosis of Alzheimer’s to support these efforts. Currently Alzheimer’s is diagnosed as a clinical entity using a process that combines cognition assessments with imaging- and spinal-fluid (“CSF”) tests. This diagnostic procedure may last for several months to a year and is usually initiated late in the disease development.
Several companies are focusing on blood as a test material. Typically, these companies employ a multi-assay strategy (multiple RNAs or proteins) combined with advanced statistical tools/algorithms to develop disease-specific diagnostic models.
Intellectual Property
We are able to protect our technology from unauthorized use by third parties only to the extent that it is covered by valid and enforceable patents or is effectively maintained as a trade secret or is protected by confidentiality agreements. Accordingly, patents or other proprietary rights are an essential element of our business. Currently, the Company does not own a patent although it does possess a license for an immunotherapy technology and two licenses for a lithium salt and proline cocrystal technology from the University of South Florida Research Foundation, Inc.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
| 16 |
While trade secret protection is an essential element of our business and we take security measures to protect our proprietary information and trade secrets, we cannot give assurance that our unpatented proprietary technology will afford us significant commercial protection. We seek to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. Our employees and consultants also sign agreements requiring that they assign to the Company their interests in intellectual property arising from their work for us. All employees sign an agreement not to engage in any conflicting employment or activity during their employment and not to disclose or misuse confidential information. However, it is possible that these agreements may be breached or invalidated, and if so, there may not be an adequate corrective remedy available. Accordingly, we cannot ensure that employees, consultants or third parties will not breach the confidentiality provisions in our contracts, infringe or misappropriate our trade secrets and other proprietary rights or that measures we take to protect our proprietary rights will be adequate.
In the future, third parties may file claims asserting that our technologies or products infringe on their intellectual property. We cannot predict whether third parties will assert such claims against us or against the licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend ourselves against such claims, whether they are with or without merit and whether they are resolved in favor of, or against, our licensors or ourselves, we may face costly litigation and the diversion of our management’s attention and resources. As a result of such disputes, we may have to develop costly non-infringing technology or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, or at all.
Alzamend currently possesses 4 service trademarks with the U.S. PTO that include its corporate name, Alzamend NeuroTM; two for its corporate slogan and one for the trade name, LiProSalTM.
Competition
Our industry is highly competitive and subject to rapid and significant technological change. While we have some, albeit limited, development experience and scientific knowledge, we will face competition from both large and small pharmaceutical and biotechnology companies, including specialty pharmaceutical companies and generic drug companies, as well as academic institutions, government agencies and research institutions, among others.
Our competition will be determined in part by the potential indications for which our products are developed and ultimately approved by regulatory authorities. It is likely that the timing of market introductions of some of our potential products or our competitors’ products will be an important competitive factor. Accordingly, the speed with which we can develop our products, conduct preclinical studies and clinical trials to obtain approval and manufacture or obtain supplies of commercial quantities of any approved products should also be important competitive factors. We expect that competition among products approved for sale will be based on additional factors, such as product efficacy, safety, reliability, availability, price and patent position.
Alzheimer’s Therapeutic Landscape
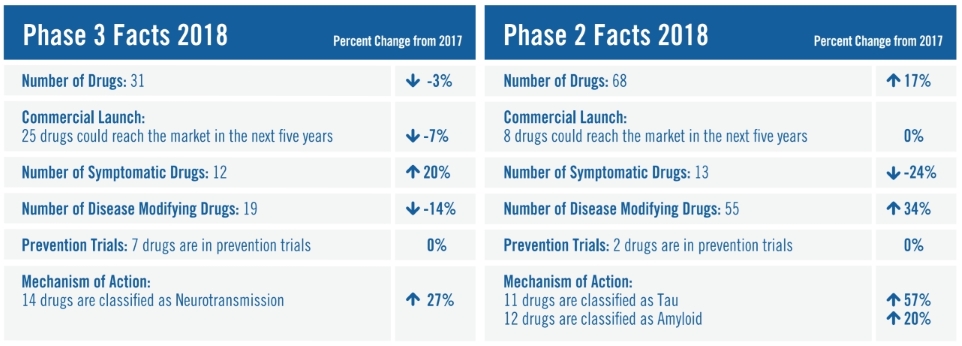
| 17 |
Current Drugs for Alzheimer’s Disease
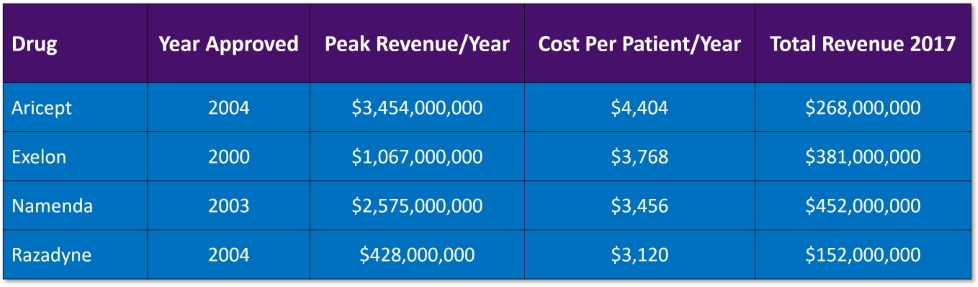
| · | Aricept – Eisai Co., Ltd. Third Quarter Financial Results (https://www.eisai.com/ir/library/settlement/pdf/e2018Q3_52.pdf). |
| · | Exelon – Novartis Pharmaceutical Co. Q4/FY 2017 Financial Report (https://www.novartis.com/sites/www.novartis.com/files/2018-01-interim-financial-report-en.pdf). |
| · | Namenda – Allergan Q4/FY 2017 Financial Report (https://www.prnewswire.com/news-releases/allergan-reports-solid-finish-to-2017-with-12-increase-in-fourth-quarter-gaap-net-revenues-to-43-billion-300593801.html). |
| · | Razadyne – Takeda FY2017 Data Book (https://www.takeda.com/siteassets/system/investors/report/quarterlyannouncements/fy2017/fy2017-full-year-results/qr2017_q4_d_en.pdf). |
| · | Thomson Reuters Report - (https://www.researchgate.net/publication/274930518_Spotlight_on_Alzheimers_disease_a_Thomson_Reuters_Pharma_Matters_report). |
Diagnostics for Alzheimer’s Disease
Cerebrospinal Fluid (CSF)
CSF samples and protein assays of particular analytes remain today the best tools in the diagnosis of Alzheimer’s disease and encephalitis. The procedure involves a lumbar puncture - the insertion of a hallow cannula or needle into the lower spinal column in order to collect 5-10 ml of blood free CSF. Until recently there have not been any in vitro diagnostic quality assays available to replace the lumbar puncture diagnostic procedure and there may not be until Saladax/Ortho Clinical Diagnostics or Roche Diagnostics release publicly their report CSF Ab42 and CSF Tau assays.
Positron Emission Tomography (PET)
PET requires large, multi-million-dollar cameras which collect the radioactive decay of minute quantities of hot radioactive tracers injected into the blood stream. The tracers emit correlated photo pairs which indicate where the tracer is staining tissue in vivo. FDG-PET is an FDA-approved tracer which measures glucose metabolism and has been successfully used to image brain energy consumption. More recently Amyvid from Avid Radiopharmaceuticals, now Lilly Diagnostics, received FDA approval as an in vivo radiotracer to label the amyloid plaques of the brain. These studies typically cost $3,000-$5,000 per imaging session per patient and require patients travel to a facility with a PET facility rather than receive a diagnostic test in their clinician’s office.
Magneto encephalography (MEG)
MEG instruments, which are both physically large and costly to facilities wishing to purchase them, employ advanced superconducting magnets operating in near absolute zero temperature to measure minute brain currents. They are scarcely available in the US and Japan, let alone any other country in the world. They are primarily used for research and will likely never become commonplace in clinical practice due to their size and cost.
| 18 |
Magnetic Resonance Imaging (MRI)
MRI instruments are able to measure the gross anatomy of the brain within the skull with resolution approaching 100 microns in a standard 1.5T clinical MRI. Although they are costly and accessible only at an imaging center (in patient or outpatient), they are standard of care to ensure that there is no gross brain tumor or evidence of white matter infarct, typical after sub-clinical or mini-strokes have occurred. In one costly modality, functional MRI is conducted whereby a patient is given tasks to complete while they are lying in an MRI brain scanner and asked to participate in task-based maneuvers to understand which anatomical structures are active during which dynamic task. These diagnostic studies are costly and difficult to implement with satisfactory results due to the distractions of motion artifacts and noise. In routine clinical practice, they are not commonly conducted.
Cognition
There are many companies creating computerized cognitive assessments of a human subject from a neuropsychological perspective. Many of these are considered reliable and easily administered in a clinician’s office. Some of the cognitive assessment tools in the market today are the CogState battery of tasks, the CNS Vital Signs, the ImPACT test and the CANTAB battery. However, these cognition assessment tools have limitations on their ability to accurately and objectively measure brain function.
Employees
As of the date of this Annual Report, the Company has two full-time and one part-time employee.
Corporate Information
Our mailing address is Alzamend Neuro, Inc., 3802 Spectrum Blvd., Suite #112C, Tampa, FL 33612 and our telephone number is (844) 722-6333. Our website address is www.alzamend.com and the www.TheAlzamendStory.com. The information contained therein or accessible thereby shall not be deemed to be incorporated into this Annual Report.
DESCRIPTION OF PROPERTY
The Company currently maintains its corporate offices at the University of South Florida Incubator Center located next to the USF Innovation Center featuring shared labs and extensive research facility.
| 19 |
RISK FACTORS
An investment in our Common Stock involves a high degree of risk. You should carefully consider the risks described below, together with all of the other information included in this Annual Report, before making an investment decision. If any of the following risks actually occurs, our business, financial condition or results of operations could suffer. In that case, the trading price of our shares of Common Stock could decline and you may lose all or part of your investment. See “Cautionary Note Regarding Forward Looking Statements” above for a discussion of forward-looking statements and the significance of such statements in the context of this Annual Report.
Risks Related to Our Company
We have virtually no operating history on which to judge our business prospects and management.
The Company was incorporated on February 26, 2016 and only commenced operations thereafter. Accordingly, we have virtually no operating history upon which to base an evaluation of our business and prospects. Operating results for future periods are subject to numerous uncertainties and we cannot assure you that the Company will achieve or sustain profitability. The Company’s prospects must be considered in light of the risks encountered by companies in the early stage of development, particularly companies in new and rapidly evolving markets. Future operating results will depend upon many factors, including our success in attracting and retaining motivated and qualified personnel, our ability to establish short term credit lines or obtain financing from other sources, such as this Offering, our ability to develop and market new products, control costs, and general economic conditions. We cannot assure you that the Company will successfully address any of these contingencies.
We are significantly influenced by our officers, directors and entities affiliated with them.
In the aggregate, beneficial ownership of the Company’s shares of Common Stock by management and affiliated parties represents approximately 55.6% of fully diluted shares of Common Stock. These stockholders, if acting together, will be able to significantly influence all matters requiring approval by stockholders, including the election of directors and the approval of mergers or other business combinations transactions.
Certain provisions of our Certificate of Incorporation could allow concentration of voting power in one stockholder, which may, among other things, delay or frustrate the removal of incumbent directors or a takeover attempt, even if such events may be beneficial to our stockholders.
Provisions of our Certificate of Incorporation adopted by our Board may delay or frustrate the removal of incumbent directors and may prevent or delay a merger, tender offer or proxy contest involving the Company that is not approved by our Board, even if those events may be perceived to be in the best interests of our stockholders. Moreover, an affiliate of our company has acquired the Series A Preferred Stock described hereinafter. Such shares have significant voting power, among other terms. Further, the Company may designate and issue separate classes of preferred stock that may entitle its holder(s) to exercise significant control over us. Consequently, anyone to whom these shares are or were issued could have sufficient voting power to significantly influence if not control the outcome of all corporate matters submitted to the vote of our common stockholders. Those matters could include the election of directors, changes in the size and composition of the Board, and mergers and other business combinations involving the Company. In addition, through any such person’s control of the Board and voting power, the affiliate may be able to control certain decisions, including decisions regarding the qualification and appointment of officers, dividend policy, access to capital (including borrowing from third-party lenders and the issuance of additional debt or equity securities), and the acquisition or disposition of assets by the Company. In addition, the concentration of voting power in the hands of an affiliate could have the effect of delaying or preventing a change in control of the Company, even if the change in control would benefit our stockholders and may adversely affect the future market price of our Common Stock should a trading market therefor develop.
Certain provisions of our Certificate of Incorporation and Bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in the stockholders’ interest.
Our Certificate of Incorporation and Bylaws and certain provisions of Delaware State law could have the effect of making it more difficult or more expensive for a third party to acquire, or discouraging a third party from attempting to acquire, control of the Company, even when these attempts may be in the best interests of our stockholders. For example, we are governed by Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. A “business combination” includes mergers, asset sales or other transactions resulting in a financial benefit to the stockholder. An “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years did own, 15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing a change in our control.
| 20 |
Limitations of Director Liability and Indemnification of Directors and Officers and Employees.
Our Certificate of Incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
| · | breach of their duty of loyalty to us or our stockholders; | |
| · | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; | |
| · | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or | |
| · | transaction from which the directors derived an improper personal benefit. |
These limitations of liability do not apply to liabilities arising under the federal or state securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our bylaws provide that we will indemnify our directors, officers and employees to the fullest extent permitted by law. Our bylaws also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding. We believe that these bylaw provisions are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability in our Certificate of Incorporation and bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
We will need but may be unable to obtain additional funding on satisfactory terms, which could dilute our stockholders or impose burdensome financial restrictions on our business.
We have relied upon cash from financing activities and in the future, we hope to rely on revenues generated from operations to fund all of the cash requirements of our activities. However, there can be no assurance that we will be able to generate any significant cash from our operating activities in the future. Future financings may not be available on a timely basis, in sufficient amounts or on terms acceptable to us, if at all. Any debt financing or other financing of securities senior to the Common Stock will likely include financial and other covenants that will restrict our flexibility. Any failure to comply with these covenants would have a material adverse effect on our business, prospects, financial condition and results of operations because we could lose our existing sources of funding and impair our ability to secure new sources of funding. However, there can be no assurance that the Company will be able to generate any investor interest in its securities. If we do not obtain additional financing, our business will never commence, in which case you would likely lose the entirety of your investment in us.
Our financial situation creates substantial doubt whether we will continue as a going concern.
Since inception, the Company has not generated revenues and has incurred losses. As of April 30, 2018, the Company had cash of $545,001 and an accumulated deficit of $2,513,137. The Company has incurred recurring losses and reported losses for the year ended April 30, 2018 of $931,663. The report of the Company’s independent registered public accounting firm on the Company’s April 30, 2018 financial statements includes a going concern explanatory paragraph which states that there is substantial doubt regarding the Company’s ability to continue as a going concern. There can be no assurances that we will be able to achieve a level of revenues adequate to generate sufficient cash flow from operations or obtain additional financing through private placements, public offerings and/or bank financing necessary to support our working capital requirements. To the extent that funds generated from any private placements, public offerings and/or bank financing are insufficient, we will have to raise additional working capital. No assurance can be given that additional financing will be available, or if available, will be on acceptable terms. If adequate working capital is not available we may be forced to discontinue operations, which would cause investors to lose their entire investment.
| 21 |
We are at an early stage of development as a company and currently have no source of revenue and may never become profitable.
We are a preclinical development stage biopharmaceutical company. Currently, we have no products approved for commercial sale and, to date, we have not generated any revenue. Our ability to generate revenue depends heavily on:
| · |
demonstration in future clinical trials that LiProSalTM and CAO22W are safe and effective; |
| · |
our ability to seek and obtain regulatory approvals, including with respect to the indications we are seeking; |
| · |
successful manufacture and commercialization of LiProSalTM and CAO22W; and |
| · |
market acceptance of LiProSalTM and CAO22W. |
We only have two product candidates, LiProSalTM and CAO22W, which are in the pre-IND stage and preclinical stage of development, respectively, and will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it and any successors could provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval and commercialize LiProSalTM or CAO22W, we will be unable to generate any revenue for many years, if at all. We do not anticipate that we will generate revenue for a few years, at the earliest, or that we will achieve profitability for at least several years after generating material revenue, if at all. If we are unable to generate revenue, we will not become profitable, and we may be unable to continue our operations.
We do not have any products that are approved for commercial sale and therefore do not expect to generate any revenues from product sales in the foreseeable future, if ever.
We currently do not have any products that are approved for commercial sale. To date, we have funded our operations primarily from loans and sales of our securities. We have not received, and do not expect to receive for at least the next few years any revenues from the commercialization of LiProSalTM or CAO22W. To obtain revenues from sales of our future product candidates, if any, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing and marketing drugs with commercial potential. We may never succeed in these activities and may not generate sufficient revenues to continue our business operations or achieve profitability.
We must effectively manage the growth of our operations, or our company will suffer.
Our initiation of operations has resulted in significantly higher operating expenses, which the net proceeds from this Offering, if any, are intended in part to offset. Expansion of our operations, to include the development of LiProSalTM and CAO22W, may also cause a significant demand on our management, finances and other resources. Our ability to manage the anticipated future growth, should it occur, will depend upon a significant expansion of our accounting and other internal management systems and the implementation and subsequent improvement of a variety of systems, procedures and controls. In addition, we intend to expand the Board and to establish an SAB. There can be no assurance that significant problems in these areas will not occur. Any failure to expand these areas and implement and improve LiProSalTM or CAO22W or our procedures and controls in an efficient manner at a pace consistent with our business could have a material adverse effect on our business, financial condition and results of operations. There can be no assurance that our attempts to expand our marketing, sales, manufacturing and customer support efforts will be successful or will result in additional sales or profitability in any future period.
Risks Related to Our Product Candidates
We have both operational and financial milestones that must be met to maintain the licensing rights to our current technology and IP from the USF Research Foundations, Inc. or those rights may be terminated.
There are certain initial license fees and milestone payments required to be paid by us to the Licensor pursuant to the terms of the License Agreements.
The License Agreement require the Company to pay royalty payments of four percent (4%) on net sales of products developed from the licensed technology for CAO22W while the License Agreements for LiProSalTM requires the Company to pay combined royalty payments of four and one-half percent (4.5%) on net sales of products developed from the licensed technology. The Company has already paid an initial license fee of $200,000 for CAO22W and $60,000 of an initial license fee of $200,000 for LiProSalTM. As an additional licensing fee for the license of CAO22W, the Licensor received 3,601,809 shares of Common Stock. As an additional licensing fee for the license of the LiProSalTM technologies, the Licensor is entitled to receive that number of shares Common Stock equal to three percent (3%) of the sum of the total number of such shares issued and outstanding shares plus any securities that are convertible into or exercisable or exchangeable for shares of Common Stock until the Company has received a total of $5 million in cash in exchange for the Company’s equity securities. Additionally, the Company is required to pay milestone payments on the due dates to the Licensor for the license of the CAO22W technology and for the LiProSalTM technologies, as follows:
| 22 |
CAO22W:
| Payment | Due Date | Event | ||||
| $ | 50,000 | September 30, 2019 | Upon IDN application filing | |||
| $ | 50,000 | 12 months from IND application filing date | Upon first dosing of patient in first Phase I Clinical Trial | |||
| $ | 175,000 | 12 months from first patient dosed in Phase I | Upon Completion of first Phase I Clinical Trial | |||
| $ | 500,000 | 24 months from completion of first Phase I Trial | Upon Completion of first Phase II Clinical Trial | |||
| $ | 1,000,000 | 12 months from completion of the first Phase II Clinical Trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA BLA Approval | |||
LiProSalTM:
| Payment | Due Date | Event | ||||
| $ | 50,000 | November 1, 2019 | Pre-IND meeting | |||
| $ | 65,000 | 6 months from IND filing date | IND application filing | |||
| $ | 190,000 | 12 months from IND filing date | Upon first dosing of patient in a clinical trial | |||
| $ | 500,000 | 12 months from first patient dosing | Upon Completion of first clinical trial | |||
| $ | 1,250,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 8 years from the Effective Date of the Agreement | Upon FDA Approval | |||
None of the milestones was met as of the date of this Annual Report. If the Company fails to meet a milestone by the specified date, the Licensor may terminate the respective License Agreement. If the Licensor were to terminate either License Agreement for whatever reason, it would materially and adversely affect our business, financial position and future prospects and you would likely lose the entirety of your investment in us.
The Licensor was also granted a preemptive right to acquire such shares or other equity securities that may be issued from time to time by the Company while the Licensor remains the owner of any equity securities of the Company. Further, if the Company issues equity securities at a price per share that is less than the price paid by purchasers in a transaction for aggregate consideration of at least $5,000,000, then the number of shares owned by the Licensor shall be increased upon such issuance. The amount of the increase shall be determined by multiplying the number of shares then owned by Licensor by a fraction; the numerator of which shall be equal to the number of shares of Common Stock outstanding immediately after the issuance of additional shares of commons stock, and the denominator of which shall be equal to the sum of (i) the number of shares of Common Stock outstanding immediately prior to the issuance of additional shares of Common Stock plus (ii) the number of shares of Common Stock which the aggregate consideration for the total number of additional shares of Common Stock so issued would purchase at the Investment Price.
If we fail to comply with our obligations in the agreements under which we license intellectual property and other rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We are a party to these License Agreements with the University and expect to enter into additional license agreements in the future. The existing License Agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty, and other obligations on us. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, we may be required to make certain payments to the licensor, we may lose the exclusivity of our license, or the licensor may have the right to terminate the license, in which event we would not be able to develop or market products covered by the license. The Licensor may take any of these actions, including terminating the License upon 60 days’ notice for any reason. Additionally, the milestone and other payments associated with these licenses will make it less profitable for us to develop our product candidates. If the Licensor were to terminate the License Agreement for whatever reason, it would materially and adversely affect our business, financial position and future prospects and you would likely lose the entirety of your investment in us.
| 23 |
In some cases, patent prosecution of our licensed technology is controlled solely by the licensor. If our licensors fail to obtain and maintain patent or other protection for the proprietary intellectual property we license from them, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. In certain cases, we control the prosecution of patents resulting from licensed technology. In the event we breach any of our obligations related to such prosecution, we may incur significant liability to our licensing partners. Licensing of intellectual property is of critical importance to our business and involves complex legal, business, and scientific issues. Disputes may arise regarding intellectual property subject to a licensing agreement, including but not limited to:
| · | the scope of rights granted under the license agreement and other interpretation-related issues; | |
| · | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; | |
| · | the sublicensing of patent and other rights; | |
| · | our diligence obligations under each of the license agreements and what activities satisfy those diligence obligations; | |
| · | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our collaborators; and | |
| · | the priority of invention of patented technology. |
If disputes over intellectual property and other rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We are substantially dependent on the success of our product candidates, which may not receive regulatory approval or be successfully commercialized.
In the future, we hope to submit LiProSalTM and CAO22W and, potentially, other product candidates, for regulatory approval. Currently, however, neither LiProSalTM nor CAO22W has been submitted for regulatory approval, which would be required before we seek to initiate commercial distribution. To date, we have invested nearly all of our resources in establishing our company and the acquisition of the intellectual property of our product candidates, LiProSalTM and CAO22W. Our near-term prospects, including our ability to finance our company and to enter into strategic collaborations and, ultimately, to generate revenue, are directly dependent upon the successful development and commercialization of LiProSalTM or CAO22W.
The development and commercial success of our product will depend on a number of factors, including, without limitation, the following:
| · | timely initiation and successful completion of preclinical studies and clinical trials for LiProSalTM or CAO22W; | |
| · | demonstration to the satisfaction of the FDA, the EMA and other applicable regulatory authorities the safety and efficacy of LiProSalTM or CAO22W as well as to obtain regulatory and marketing approval for LiProSalTM or CAO22W in the U.S., Europe and elsewhere; | |
| · | continued compliance with all clinical and regulatory requirements applicable to LiProSalTM and CAO22W; | |
| · | maintenance of an acceptable safety profile of LiProSalTM and CAO22W following regulatory approval; | |
| · | competition with other treatments; | |
| · | creation, maintenance and protection of our intellectual property portfolio, including patents and trade secrets, and regulatory exclusivity for LiProSalTM and CAO22W; | |
| · | effectiveness of our and our eventual partners’ marketing, sales and distribution strategy and operations; | |
| · | ability of our third-party manufacturers to manufacture supplies of our product and product candidates and to develop, validate and maintain commercially viable manufacturing processes; | |
| · | ability to launch commercial sales of LiProSalTM or CAO22W following regulatory approval, whether alone or in collaboration with others; and | |
| · | acceptance of LiProSalTM or CAO22W from physicians, health care payers, patients and the medical community. |
| 24 |
Many of these factors are beyond our control, and we cannot assure you that we will ever be able to generate sufficient revenue or any revenue from the sale of LiProSalTM or CAO22W. Our failure in any of the above factors, or in successfully commercializing LiProSalTM or CAO22W on a timely basis, could have a material adverse effect on our business, results of operations and financial condition, and the value of your investment could substantially decline.
LiProSalTM and CAO22W may not achieve market acceptance, which would limit our ability to generate revenue from new products.
Even if we develop LiProSalTM or CAO22W and gain regulatory approvals for either or both, unless physicians and patients accept our product candidates, we may not be able to sell them and generate significant revenues. We cannot assure you that LiProSalTM, CAO22W, or any other potential products, will achieve market acceptance and revenue if and when they obtain the requisite regulatory approvals. Market acceptance of any product candidate depends on a number of factors, including but not limited to:
| · | the indication and warnings approved by regulatory authorities in the product label; | |
| · | continued demonstration of efficacy and safety in commercial use; | |
| · | physicians’ willingness to prescribe the product; | |
| · | reimbursement from third-party payors such as government health care systems and insurance companies; | |
| · | the price of the product; | |
| · | the nature of any post-approval risk management plans mandated by regulatory authorities; | |
| · | competition; and | |
| · | the effectiveness of marketing and distribution support. |
Any failure by LiProSalTM or CAO22W to achieve market acceptance or commercial success could have a material adverse effect on our business, results of operations and financial condition.
Problems in our manufacturing process, failure to comply with manufacturing regulations or unexpected increases in our manufacturing costs could harm our business, results of operations and financial condition.
We are responsible for the manufacture and supply of LiProSalTM and CAO22W, independently of each other. The manufacturing of LiProSalTM and CAO22W necessitates compliance with the FDA, EU and international current Good Manufacturing Practice (“cGMP”) and other international regulatory requirements. Although we may in the future contract with third parties for a certain amount of the manufacturing of LiProSalTM and CAO22W, the responsibility to obtain market authorization for LiProSalTM and CAO22W remains with us. As such, even if we could potentially have a claim against one or more third parties, we are legally liable for any noncompliance related to LiProSalTM and CAO22W and we expect to retain legal responsibility for any future product candidates as well.
If we are unable to manufacture, or contract to manufacture, LiProSalTM and CAO22W in accordance with regulatory specifications, or if there are disruptions in the manufacturing process due to damage, loss or failure to pass regulatory inspections of manufacturing facilities, we may not be able to meet the demand for our products or supply sufficient product for use in clinical trials, and this may harm our ability to commercialize LiProSalTM and CAO22W on a timely or cost-competitive basis, or preclude us from doing so at all.
Before we can begin commercial manufacture of LiProSalTM, CAO22W, or any other product candidate that we may develop in the future, we must obtain FDA regulatory approval for the product, which requires a successful FDA inspection of our manufacturing facilities, processes and quality systems in addition to other product-related approvals. Even if we successfully pass an FDA Pre-Approval Inspection of any manufacturing facilities we may establish or contract with, our pharmaceutical facilities would be continuously subject to inspection by the FDA and foreign regulatory authorities, even after product approval. Due to the complexity of the processes that we anticipate will eventually be used to manufacture LiProSalTM and CAO22W, we may be unable to pass federal, state or international regulatory inspections in a cost-effective manner, whether initially or at any time thereafter. If we are unable to comply with manufacturing regulations, we may be subject to fines, unanticipated compliance expenses, recall or seizure of any approved products, or legal actions such as injunctions or criminal or civil prosecution. These possible sanctions could materially and adversely affect our business, results of operations and financial condition. See also “Risks Related to Development and Regulatory Approval of Our Product.” The regulatory approval process is uncertain, requires us to utilize significant resources, and may prevent us or our commercial partners from obtaining approvals for the commercialization of some or all of our drug candidates.”
| 25 |
We expect to face substantial competition, which may result in others discovering, developing or commercializing products before, or more successfully than, we do.
The development and commercialization of new therapy and vaccine products is highly competitive. We will face competition with respect to LiProSalTM, CAO22W and any other product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. In addition to existing therapeutic treatments for the indications we are targeting with LiProSalTM and CAO22W, we also face potential competition from other drug candidates in development by other companies. Our potential competitors include large health care companies, such as Celgene Corporation, Merck & Co., Inc., Sanofi S.A., Eli Lilly and Company, Bayer AG, Novartis AG and Boehringer Ingelheim GmbH. We also know of several smaller early stage companies that are developing products for use in our segment of the market. Some of the potential competitive compounds referred to above are being developed by large, well-financed and established pharmaceutical and biotechnology companies or have been partnered with such companies, which may give them development, regulatory and marketing advantages over our products.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third-party payers seeking to encourage the use of generic products. If LiProSalTM or CAO22W achieves marketing approval, we expect that it will be priced at a significant premium over competing generic products.
Some of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we are unable to compete successfully, we may be unable to grow and sustain our revenue, which could materially and adversely affect our business, results of operations and financial condition.
Serious adverse events or other safety risks could require us to abandon development and preclude, delay or limit approval of LiProSalTM or CAO22W, or limit the scope of any approved label or market acceptance.
If LiProSalTM, CAO22W or any other product candidate that we may develop in the future, prior to or after any approval for commercial sale, causes serious or unexpected side effects, or become associated with other safety risks such as misuse, abuse or diversion, a number of potentially significant negative consequences could result, including, without limitation:
| · | regulatory authorities may interrupt, delay or halt clinical trials; | |
| · | regulatory authorities may deny regulatory approval of LiProSalTM or CAO22W; | |
| · | regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, or impose restrictions on distribution in the form of a Risk Evaluation and Mitigation Strategy “REMS”), in connection with approval, if any; | |
| · | regulatory authorities may withdraw their approval, require more onerous labeling statements or impose a more restrictive REMS of any product that is approved; | |
| · | we may be required to change the way the product is administered or conduct additional clinical trials; | |
| · | any relationships that we may be able to form in the future with any commercial partners may suffer; | |
| · | we could be sued and held liable for harm caused to patients; and | |
| · | our reputation may suffer. |
We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants or if preliminary data demonstrate that either CAO22W or LiProSalTM are unlikely to receive regulatory approval or is unlikely to be successfully commercialized. In addition, regulatory agencies, an Institutional Review Board, or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate a clinical trial of LiProSalTM, CAO22W or any other product candidate that we may in the future develop, the commercial prospects for that product will be harmed and our ability to generate product revenue from that product may be delayed or eliminated. Furthermore, any of these events could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing LiProSalTM or CAO22W and materially impair our ability to generate revenue from the commercialization of LiProSalTM or CAO22W either by us or by any commercial partners that we may develop a relationship with in the future and could have a material adverse effect on our reputation, business, results of operations and financial condition.
| 26 |
If we fail to obtain and sustain an adequate level of reimbursement for our products by third-party payers, sales and profitability will be adversely affected.
The course of medical treatment for human patients is, and will continue to be, expensive. We expect that most patients and their families will not be capable of paying for our products themselves. Accordingly, it is unlikely that there will be a commercially viable market for LiProSalTM or CAO22W without reimbursement from third-party payers. Additionally, even if there is a commercially viable market, if the level of third-party reimbursement is insufficient from the patient’s perspective, our revenue and gross margins will be materially and adversely affected.
A current trend in the U.S. health care industry, as well as in other countries around the world, is toward cost containment. Large public and private payers, managed care organizations, group purchasing organizations and similar organizations are exerting increasing influence on decisions regarding the use of, and reimbursement levels for, particular treatments. Third-party payers, such as government programs, including Medicare in the U.S., and private health care insurers, carefully review and have increasingly been challenging the coverage of, and prices charged for, medical products and services. Many third-party payers limit coverage of or reimbursement for newly-approved health care products. Reimbursement rates from private health insurance companies vary depending on the company, the insurance plan and other factors. Cost-control initiatives could decrease the price we or our partners establish for products, which could result in lower product revenue and profitability.
Reimbursement systems in international markets vary significantly by country and by region, and reimbursement approvals must be obtained on a country-by-country basis. Our eventual partners may elect to reduce the price of our products in order to increase the likelihood of obtaining reimbursement approvals. In many countries, products cannot be commercially launched until reimbursement is approved and the negotiation process in some countries can exceed 12 months. In addition, pricing and reimbursement decisions in certain countries can be affected by decisions taken in other countries, which can lead to mandatory price reductions and/or additional reimbursement restrictions across a number of other countries, which may adversely affect our sales and profitability. If countries set prices that are not sufficient to allow us or our partners to generate a profit, our partners may refuse to launch the product in such countries or withdraw the product from the market, which would adversely affect our sales and profitability and could materially and adversely affect our business, results of operations and financial condition.
We may not be successful in our efforts to expand our pipeline of product candidates.
One element of our strategy is to expand our pipeline of pharmaceuticals based on our technology and advance these product candidates through clinical development for the treatment of a variety of indications. Although our research and development efforts to date have resulted in a number of development programs based on our technology, we may not ultimately be able to develop product candidates that are safe and effective. Even if we are successful in continuing to expand our pipeline, the potential product candidates that we identify may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to receive marketing approval and achieve market acceptance. In addition, if we attempt to apply our technology to develop product candidates for indications outside of Alzheimer’s, we will need to conduct genotoxicity and immunotoxicity trials, in which the results are presently uncertain. If we do not successfully develop and commercialize product candidates based upon our technological approach, we will not be able to obtain product revenue in future periods, which would make it unlikely that we would ever achieve profitability.
| 27 |
Product recalls or inventory losses caused by unforeseen events, cold chain interruption and testing difficulties may adversely affect our operating results and financial condition.
LiProSalTM and CAO22W, individually, will be manufactured and distributed, if ever, using technically complex processes requiring specialized facilities, highly specific raw materials and other production constraints. The complexity of these processes, as well as the strict company and government standards for the manufacture of our products, will subject us to production risks. While product batches released for use in clinical trials or for commercialization undergo sample testing, some defects may only be identified following product release. In addition, process deviations or unanticipated effects of approved process changes may result in these intermediate products not complying with stability requirements or specifications. Most of our products must be stored and transported at temperatures within a certain range, which is known as “strict cold chain” storage and transportation. If these environmental conditions deviate, our products’ remaining shelf lives could be impaired or their efficacy and safety could become adversely affected, making them no longer suitable for use. The occurrence or suspected occurrence of production and distribution difficulties can lead to lost inventories, and in some cases product recalls, with consequential reputational damage and the risk of product liability. The investigation and remediation of any identified problems can cause production delays, substantial expense, lost sales and delays of new product launches, any of which could have a material adverse effect on our business, results of operations and financial condition.
| 28 |
Risks Related to Development and Regulatory Approval of Our Product
There is a high rate of failure for drug candidates proceeding through clinical trials.
Generally speaking, there is a high rate of failure for drug candidates proceeding through clinical trials. We may suffer significant setbacks in our clinical trials similar to the experience of a number of other companies in the pharmaceutical and biotechnology industries, even after receiving promising results in earlier trials. Further, even if we view the results of a clinical trial to be positive, the FDA or other regulatory authorities may disagree with our interpretation of the data. For instance, any such differing interpretation could cause the FDA to require additional trials. In the event that:
| · |
we obtain negative results from the LiProSalTM or CAO22W from a clinical trial;
| |
| · |
the FDA places a clinical hold on our clinical trials due to potential chemistry, manufacturing and controls issues or other hurdles, or
| |
| · |
the FDA does not approve our NDA for LiProSalTM or our BLA for CAO22W, then: |
| o | we may not be able to generate sufficient revenue or obtain financing to continue our operations; | |
| o |
our ability to execute our current business plan will be materially impaired;
| |
| o |
our reputation in the industry and in the investment community would likely be significantly damaged, and
| |
| o | the price of the Common Stock, assuming a trading market has then developed therefor, would likely decrease significantly. |
Any of these results could materially and adversely affect our business, results of operations or financial condition.
Clinical trials for LiProSalTM or CAO22W can be expensive, time consuming, uncertain and susceptible to change, delay or termination.
Clinical trials are expensive, time consuming and difficult to design and implement. The result of a clinical trial may be undesirable and can result in a clinical trial cancellation or the need for re-evaluation and supplementation. Even if the results of our clinical trials are favorable, the clinical trials for LiProSalTM or CAO22W are expected to continue for a few years and may even take significantly longer to complete. In addition, we, the FDA, an IRB, or other regulatory authority, including in the U.S., EU and elsewhere, may suspend, delay or terminate our clinical trials at any time, for various reasons, including, without limitation:
| · |
lack of effectiveness of LiProSalTM or CAO22W during clinical trials;
| |
| · |
discovery of serious or unexpected toxicities or side effects experienced by trial participants or other safety issues;
| |
| · |
slower than expected rates of subject recruitment and enrollment rates in clinical trials;
| |
| · |
difficulty in retaining subjects who have initiated a clinical trial but may have withdrawn due to adverse side effects from the therapy, insufficient efficacy, fatigue with the clinical trial process or for any other reason;
| |
| · |
delays or inability in manufacturing or obtaining sufficient quantities of materials for use in clinical trials due to manufacturing or regulatory constraints;
| |
| · |
inadequacy of or changes in our manufacturing process or product formulation;
| |
| · |
delays in obtaining regulatory authorization to commence a trial, including experiencing “clinical holds” or delays requiring suspension or termination of a trial by a regulatory agency, such as the FDA, before or after a trial is commenced;
| |
| · |
changes in applicable regulatory policies and regulations;
|
| 29 |
| · |
delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective clinical trial sites;
| |
| · |
delay or failure to supply product for use in clinical trials which conforms to regulatory specification;
| |
| · |
unfavorable results from ongoing pre-clinical studies and clinical trials;
| |
| · |
failure of any contract research organizations (“CROs”) that we may partner with in the future, or other third-party contractors, to comply with all contractual requirements or to perform their services in a timely or acceptable manner;
| |
|
|
· |
failure by us, our employees, any CROs or their employees to comply with all applicable FDA or other regulatory requirements relating to the conduct of clinical trials;
|
| · |
scheduling conflicts with participating clinicians and clinical institutions;
| |
| · |
failure to design appropriate clinical trial protocols; or
| |
| · | regulatory concerns with pharmaceutical products generally and the potential for abuse. |
Any of the foregoing could have a material adverse effect on our business, results of operations and financial condition.
The regulatory approval process is uncertain, requires us to utilize significant resources, and may prevent us or our commercial partners from obtaining approvals for the commercialization of LiProSalTM or CAO22W.
The research, testing, manufacturing, labeling, approval, sale, marketing and testing of LiProSalTM and CAO22W are and will be subject to extensive regulation by regulatory authorities in the U.S., Europe and elsewhere, and regulatory requirements applicable to our product differ from country to country. Neither we nor any commercial partner is permitted to market any of our current or future product candidates in the U.S. until we receive approval from the FDA of either an NDA or BLA, respectively. Obtaining approval of an NDA or a BLA can be an uncertain process that requires us to utilize significant resources. Furthermore, regulatory authorities possess broad discretion regarding processing time and usually request additional information and raise questions which have to be answered. There is considerable uncertainty regarding the times at which products may be approved. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including: warning letters, civil and criminal penalties, injunctions, withdrawal of approved products from the market, product seizure or detention, product recalls, total or partial suspension of production, and refusal to approve pending applications or supplements to approved applications.
The process required by the FDA and most foreign regulatory authorities before human health care pharmaceuticals may be marketed generally involves nonclinical laboratory and, in some cases, animal tests; submission of an IND, which must become effective before clinical trials may begin; adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use or uses; pre-approval inspection of manufacturing facilities and clinical trial sites; and FDA approval of a NDA or BLA, which must occur before a drug can be marketed or sold.
Regulatory approval of an NDA or BLA, or any supplement thereof, is not guaranteed, and the approval process requires us to utilize significant resources, could take several years, and is subject to the substantial discretion of the FDA. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon or have to repeat or perform additional studies. If our product or any of our future product candidates fails to demonstrate safety and efficacy in our studies, or for any other reason does not gain regulatory approval, our business and results of operations will be materially and adversely harmed.
In addition, separate regulatory approvals are required in order to market any product in many jurisdictions, including the U.S., the European Economic Area, which consists of the 28 Member States of the European Union plus Norway, Iceland and Liechtenstein, and many others. Approval procedures vary among countries and can involve additional studies and testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Studies conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may be unable to file for regulatory approvals or do so on a timely basis and, even if we are able to, we may not receive necessary approvals to commercialize our products in any market. Any of these results could have a material adverse effect on our business, results of operations and financial condition.
| 30 |
Even if we receive regulatory approval for any of our future product candidates, we will be subject to ongoing FDA and other regulatory body obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if any, if approved, will be subject to labeling and manufacturing requirements and could be subject to other restrictions. Failure to comply with these regulatory requirements or the occurrence of unanticipated problems with our products could result in significant penalties.
Any regulatory approvals that we or any of our collaborators receive for LiProSalTM, CAO22W or any future product candidate may be subject to conditions of approval or limitations on the approved indicated uses for which the product may be marketed or may contain requirements for potentially costly surveillance to monitor the safety and efficacy of the product candidate. In addition, LiProSalTM, CAO22W and any of our future product candidates, if approved by the FDA or other regulatory bodies, will be subject to extensive and ongoing regulatory requirements regarding the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping. These requirements will include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP, Good Laboratory Practice and Good Clinical Practice for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| · |
restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls;
| |
| · |
fines, warning letters or holds on target studies;
| |
| · |
refusal by the FDA or other applicable regulatory body to approve pending applications or supplements to approved applications filed by us or our strategic collaborators, or suspension or revocation of product license approvals;
| |
| · |
product seizure or detention, or refusal to permit the import or export of products; and
| |
| · | injunctions or the imposition of civil or criminal penalties. |
The policies of the FDA and other regulatory bodies may change, and additional government regulations may be promulgated that could prevent, limit or delay regulatory approval of LiProSalTM or CAO22W. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or elsewhere. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability, which would materially and adversely affect our business, results of operations and financial condition.
LiProSalTM or CAO22W and any of our future product candidates, if approved, may cause or contribute to adverse medical events that we are required to report to the FDA and regulatory authorities in other countries and, if we fail to do so, we could be subject to sanctions that would materially harm our business.
If we are successful in commercializing LiProSalTM, CAO22W or any of our future product candidates, regulations promulgated by the FDA and by the regulatory authorities in other countries require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA and regulatory authorities in other countries could take action including criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval or clearance of future products, which could have a material adverse effect on our business, results of operations and financial condition.
| 31 |
Legislative or regulatory reforms with respect to products may make it more difficult and costly for us to obtain regulatory clearance or approval of LiProSalTM, CAO22W or any of our future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress and lawmaking bodies in other countries that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Similar changes in regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the U.S. or in other countries may impose additional costs or lengthen review times of LiProSalTM, CAO22W and any of our future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| · |
requests for additional endpoints or studies;
| |
| · |
changes to manufacturing methods;
| |
| · |
recall, replacement, or discontinuance of certain products; and
| |
| · | additional record keeping. |
Each of these would likely entail substantial time and cost and could have a material adverse effect on our ability to obtain regulatory approval for our product candidates. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products could materially and adversely affect our business, results of operations and financial condition.
Our ability to market LiProSalTM, CAO22W and any future product candidates in the U.S., if approved, will be limited to use for the treatment of the indications for which they are approved, and if we want to expand the indications for which we may market LiProSalTM, CAO22W and any future product candidates, we will need to obtain additional FDA approvals, which may not be granted.
We plan to seek full FDA approval in the U.S. for LiProSalTM and CAO22W to treat Alzheimer’s disease. If LiProSalTM or CAO22W is approved, the FDA will restrict our ability to market or advertise it for the treatment of indications other than the one for which it is approved, which could limit its use. If we decide to attempt to develop, promote and commercialize new treatment indications and protocols for LiProSalTM, CAO22W and product candidates in the future, we could not predict when, or if, we would ever receive the approvals required to do so. We would be required to conduct additional studies to support such applications for additional use, which would consume additional resources and may produce results that do not result in FDA approvals. If we do not obtain additional FDA approvals, our ability to expand our business in the U.S. would be adversely affected, which could materially and adversely affect our business, results of operations and financial condition.
The anticipated development of a REMS for LiProSalTM or CAO22W could cause delays in the approval process and would add additional layers of regulatory requirements that could impact our ability to commercialize LiProSalTM and CAO22W in the U.S. and reduce their market potential.
As a condition of approval of an NDA or a BLA, the FDA may require a REMS to ensure that the benefits of the drug outweigh the potential risks. REMS elements can include medication guides, communication plans for health care professionals, and elements to assure safe use (“ETASU”). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. We may be required to adopt a REMS for LiProSalTM or CAO22W to ensure that the benefits outweigh the risks of abuse, misuse, diversion and other potential safety concerns. Even if the risk of abuse, misuse or diversion are not as high as for some other products, there can be no assurance that the FDA will approve a manageable REMS for LiProSalTM or CAO22W, which could create material and significant limits on our ability to successfully commercialize LiProSalTM and CAO22W in the U.S. Delays in the REMS approval process could result in delays in the NDA or BLA approval process, respectively. In addition, as part of the REMS, the FDA could require significant restrictions, such as restrictions on the prescription, distribution and patient use of the product, which could significantly impact our ability to effectively commercialize LiProSalTM or CAO22W, and dramatically reduce their market potential thereby adversely impacting our business, financial condition and results of operations. Even if initial REMS are not highly restrictive, if, after launch, LiProSalTM, CAO22W and other drug candidates were to be subject to significant abuse/non-medical use or diversion from licit channels, this could lead to negative regulatory consequences, including a more restrictive REMS, which could materially and adversely affect our business, results of operations and financial condition.
| 32 |
If we are found in violation of “fraud and abuse” laws, we may be required to pay a penalty and/or be suspended from participation in government-run health care programs, which may adversely affect our business, financial condition and results of operations.
If we are successful in obtaining marketing approval for our products in the U.S. and elsewhere, we will be subject to various health care “fraud and abuse” laws, including anti-kickback laws, false claims laws and other laws intended to reduce fraud and abuse in government-run health care programs, which could affect us, particularly upon successful commercialization of our products in the U.S. For example, the Medicare and Medicaid Patient Protection Act of 1987 (otherwise known as the federal “Anti-Kickback Statute”) makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce the referral of business, including the purchase, order or prescription of a particular drug for which payment may be made under a U.S. health care program such as Medicare or Medicaid. Under U.S. federal government regulations, some arrangements, known as safe harbors, are deemed not to violate the Anti-Kickback Statute. Although we intend to seek to structure our business arrangements in compliance with all applicable requirements, these laws are broadly written, and it is often difficult to determine precisely how the law will be applied in specific circumstances. Accordingly, it is possible that our practices may be challenged under the Anti-Kickback Statute and similar laws in other jurisdictions. False claims laws prohibit anyone from knowingly and willfully presenting or causing to be presented for payment to third-party payers, including government payers, reimbursement claims for drugs or services that are false or fraudulent, claims for items or services that were not provided as claimed, or claims for medically unnecessary items or services. Cases have been brought under false claims laws alleging that off-label promotion of pharmaceutical products or the payment of kickbacks to pharmaceutical providers has resulted in the submission of false claims to governmental health care programs. Under laws such as the Health Insurance Portability and Accountability Act of 1996 in the U.S., we are prohibited from knowingly and willfully executing a scheme to defraud any health care benefit program, including private payers, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and/or exclusion or suspension from government-run health care programs such as Medicare and Medicaid and debarment from contracting with the U.S. and other governments. In addition, in the U.S. individuals have the ability to bring actions on behalf of the government under the federal False Claims Act as well as under state false claims laws.
Many states in the U.S. have adopted laws similar to the Anti-Kickback Statute, some of which apply to the referral of patients for health care services reimbursed by any source, not just governmental payers. In addition, California and a few other states in the U.S. have passed laws that require pharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America Code on Interactions with Health Care Professionals. In addition, several states impose other marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to the state. There are ambiguities as to what is required to comply with these state requirements and if we fail to comply with an applicable state law requirement we could be subject to penalties.
We have yet to receive definitive guidance on the application of fraud and abuse laws to our business. Law enforcement authorities are increasingly focused on enforcing these laws, and it is possible that some of our future practices may be challenged under these laws. While we believe we will be able to structure our business arrangements to comply with these laws, it is possible that the government could in the future allege violations of, or convict us of violating, these laws. If we are found in violation of one of these laws, we could be required to pay a penalty and could be suspended or excluded from participation in certain government-run health care programs, and our business, results of operations and financial condition may be materially and adversely affected.
| 33 |
Risks Related to Our Business and Industry
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop LiProSalTM, CAO22W or any future product candidates, conduct our in-licensing and development efforts or commercialize LiProSalTM, CAO22W or any of our future product candidates.
Our future growth and success depends in part on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Stephan Jackman, our Chief Executive Officer, Kenneth Cragun, our Chief Financial Officer as well as on Dr. Chuanhai Cao, the neuroscientist who developed CAO22W, as well as the senior scientists on Dr. Cao’s medical research team, Dr. Roland “Doug” Shytle and Dr. Jun Tan, two of the investors of LiProSalTM and other members of our senior management team. The loss of services of any of these individuals could delay or prevent the successful development of our current or future product pipeline, completion of our planned development efforts or the commercialization of LiProSalTM or CAO22W. It is possible that current or former employees of the Company could put forward claims for an alleged right to our patents and demand compensation therefor. If one or more of the key personnel were to leave us and engage in competing operations, our business, results of operations and financial condition could be materially and adversely affected. To date, none of our key personnel has left us or, to our knowledge, engaged in competing operations, nor has any departure of key personnel had any material effect on our company.
We may have trouble hiring additional qualified personnel.
As we expand our development and commercial activities, we will need to hire additional personnel and could experience difficulties attracting and retaining qualified employees. Competition for qualified personnel in the biopharmaceutical field is intense due to the limited number of individuals who possess the skills and experience required by that industry. We may not be able to attract and retain quality personnel on favorable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that such personnel have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output. Any of these difficulties could have a material adverse effect on our business, results of operations and financial condition.
We are subject to risks relating to legal proceedings.
We are subject to various claims and legal actions arising in the ordinary course of our business. Any such litigation could be very costly and could distract our management from focusing on operating our business. The existence of any such litigation could harm our business, results of operations and financial condition. Results of actual and potential litigation are inherently uncertain. An unfavorable result in a legal proceeding could adversely affect our reputation, financial condition and operating results.
If product liability lawsuits are brought against us, we will incur substantial liabilities and may be required to limit the commercialization of LiProSalTM or CAO22W.
We and our partners face potential product liability exposure related to the testing of LiProSalTM or CAO22W in clinical trials. We will face exposure to claims by an even greater number of persons if we begin to market and distribute our products commercially in the U.S. and elsewhere, including those relating to misuse of LiProSalTM or CAO22W. Now, and in the future, an individual may bring a liability claim against us alleging that LiProSalTM or CAO22W caused an injury. While we intend to take what we believe to be appropriate precautions, we may be unable to avoid significant liability if any product liability lawsuit is brought against us. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities. Even if we successfully defend any such action, the costs associated with such defense could prove exorbitant. Regardless of merit or eventual outcome, liability claims may result in:
| · |
decreased demand for LiProSalTM or CAO22W, if such product candidate is approved;
| |
| · |
injury to our reputation;
| |
| · |
withdrawal of clinical trial participants;
| |
| · |
costs of related litigation;
| |
| · |
substantial monetary awards to patients and others;
| |
| · |
increased cost of liability insurance;
| |
| · |
loss of revenue; and
| |
| · | our inability to successfully commercialize our products. |
| 34 |
Furthermore, in the future there may be a need to expand the scope of our insurance coverage, which could result in significantly increased costs or the inability to obtain sufficient insurance coverage. Any of these occurrences could have a material adverse effect on our business, results of operations and financial condition.
Failure of our information technology systems could significantly disrupt the operation of our business.
Our ability to execute our business plan and to comply with regulatory requirements with respect to data control and data integrity depends, in part, on the continued and uninterrupted performance of our information technology systems (“IT systems”). These systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, there are no assurances that electronic break-ins, computer viruses and similar disruptive problems, and/or sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data will not occur. The occurrence of any of the foregoing with respect to our IT systems could have a material adverse effect on our business, results of operations or financial condition.
We will be subject to the U.S. Foreign Corrupt Practices Act and other anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our anticipated operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations, if initiated, will be subject to certain anti-corruption laws, including the U.S. Foreign Corrupt Practices Act (“FCPA”), and other anti-corruption laws that apply in countries where we do business. The FCPA and other anti-corruption laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and our commercial partners operate in a number of jurisdictions that pose a high risk of potential FCPA violations and we participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We also anticipate becoming subject to other laws and regulations governing our international operations, including regulations administered in the U.S. and in the EU, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations (collectively, “Trade Control Laws”).
There can be no assurance that we will be completely effective in ensuring our compliance with all applicable anticorruption laws, including the FCPA or other legal requirements, such as Trade Control Laws. Any investigation of potential violations of the FCPA, other anti-corruption laws or Trade Control Laws by U.S., EU or other authorities could have an adverse impact on our reputation, our business, results of operations and financial condition. Furthermore, should we be found not to be in compliance with the FCPA, other anti-corruption laws or Trade Control Laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, as well as the accompanying legal expenses, any of which could have a material adverse effect on our reputation and liquidity, as well as on our business, results of operations and financial condition.
Risks Related to Our Intellectual Property
We may be forced to litigate to enforce or defend our intellectual property rights, or the intellectual property rights of our licensors.
We may be forced to litigate to enforce or defend our intellectual property rights against infringement and unauthorized use by competitors. In so doing, we may place our intellectual property at risk of being invalidated, held unenforceable, or narrowed in scope. Further, an adverse result in any litigation or defense proceedings may place pending applications at risk of non-issuance. In addition, if any licensor fails to enforce or defend its intellectual property rights, this may adversely affect our ability to develop and commercialize LiProSalTM or CAO22W as well as our ability to prevent competitors from making, using, and selling competing products. Any such litigation could be very costly and could distract our management from focusing on operating our business. The existence or outcome of any such litigation could harm our business, results of operations and financial condition.
| 35 |
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential and proprietary information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our Common Stock, should a market therefor ever develop.
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We rely on trade secrets to protect our proprietary know-how and technological advances, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain trade secret protection or failure to adequately protect our intellectual property could enable competitors to develop generic products or use our proprietary information to develop other products that compete with our products or cause additional, material adverse effects upon our business, results of operations and financial condition.
The transfer of technology and knowledge to contract manufacturers pursuant to the production of our products also creates a risk of uncontrolled distribution and copying of concepts, methods and processes relating to our products. Such uncontrolled distribution and copying could have a material adverse effect on the value of our products if used for the production of competing drugs or otherwise used commercially without our obtaining financial compensation.
We may become subject to third parties’ claims alleging infringement of patents and proprietary rights or seeking to invalidate our patents or proprietary rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of LiProSalTM or CAO22W.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry, as well as patent challenge proceedings, including interference and administrative law proceedings before the U.S. Patent and Trademark Office (“U.S. PTO”) and the European Patent Office (“EPO”), and oppositions and other comparable proceedings in other jurisdictions. Recently, under U.S. patent reform laws, new procedures including inter partes review and post grant review have been implemented. As stated below, the novel implementation of such reform laws presents uncertainty regarding the outcome of challenges to our patents in the future.
We cannot assure you that LiProSalTM, CAO22W or any of our future product candidates will not infringe existing or future patents. We may be unaware of patents that have already issued that a third party might assert are infringed by LiProSalTM, CAO22W or one of our future product candidates. Because patent applications can take many years to issue and may be confidential for eighteen months or more after filing, there may be applications now pending of which we are unaware of and which may later result in issued patents that we may infringe by commercializing LiProSalTM, CAO22W or any of our future product candidates. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Moreover, we may face claims from non-practicing entities (commonly referred to as patent trolls), which have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
We may be subject to third-party claims in the future against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing a third party’s patents. If a patent infringement suit were brought against us or our collaborators, we or our collaborators could be forced to stop or delay research, development, manufacturing or sales of LiProSalTM or CAO22W. As a result of patent infringement claims, or in order to avoid potential claims, we or our collaborators may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or forced to redesign it, or to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. Even if we are successful in defending such claims, infringement and other intellectual property litigation can be expensive and time-consuming to litigate and divert management’s attention from our core business. Any of these events could harm our business significantly.
| 36 |
In addition to infringement claims against us, if third parties have prepared and filed patent applications in the U.S. that also claim technology to which we have rights, we may have to participate in interference proceedings in the U.S. PTO to determine the priority of invention. Third parties may also attempt to initiate reexamination, post grant review or inter partes review of our patents in the U.S. PTO. We may also become involved in similar opposition proceedings in the EPO or comparable offices in other jurisdictions regarding our intellectual property rights with respect to our products and technology. Any of these claims could have a material adverse effect on our business, results of operations and financial condition.
If our efforts to protect the proprietary nature of the intellectual property related to LiProSalTM, CAO22W or any of our potential future product candidates are not adequate, we may not be able to compete effectively in our market.
We expect to rely upon a combination of patents, trade secret protection as well as confidentiality and license agreements to protect the intellectual property related to our product and our current product candidates and our development programs.
Composition-of-matter patents on an active pharmaceutical ingredient are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, as such patents provide protection without regard to any particular method of use or manufacture. We cannot be certain that the claims in any patent application that we may submit covering composition-of-matter of LiProSalTM, CAO22W and any potential future product candidates will be considered patentable by the U.S. PTO and courts in the U.S., or by the patent offices and courts in foreign countries. Method-of-use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method.
The strength of patents involves complex legal and scientific questions and can be uncertain. The patent applications that we may in the future own or license may fail to result in issued patents in the U.S. or in other foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, any of our future patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the patent applications we may in the future own, in-license or pursue with respect to LiProSalTM, CAO22W or any future product candidates is threatened, it could threaten our ability to commercialize LiProSalTM, CAO22W or any future product candidates. Further, if we encounter delays in our development efforts, the period of time during which we could market LiProSalTM, CAO22W or any future product candidates under patent protection would be reduced. Since patent applications in the U.S. and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to LiProSalTM, CAO22W, or any future product candidates.
Even where laws provide protection, costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have.
We will also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, nor that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the EU or the U.S. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the U.S. and elsewhere. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially and adversely affect our business, results of operations and financial condition.
Any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
| 37 |
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect LiProSalTM and CAO22W.
As is the case with other biopharmaceutical companies, our success will be heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in other situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the U.S. PTO, the laws and regulations governing patents could change in ways that would weaken our ability to obtain patents and to enforce patents that we might obtain in the future. Similarly, changes in EU patent law and elsewhere could negatively affect the value of our patents registered outside of the U.S.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with any of these requirements.
The U.S. PTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case, which could have a material adverse effect on our business, results of operations and financial condition.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on LiProSalTM, CAO22W and any future product candidates throughout the world is prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
| 38 |
Item 2. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of our operations together with our financial statements and the notes thereto appearing elsewhere in this Annual Report. This discussion contains forward-looking statements reflecting our current expectations, whose actual outcomes involve risks and uncertainties. Actual results and the timing of events may differ materially from those stated in or implied by these forward-looking statements due to a number of factors, including those discussed in the sections entitled “Risk Factors,” “Cautionary Statement regarding Forward-Looking Statements” and elsewhere in this Annual Report. Please see the notes to our Financial Statements for information about our Significant Accounting Policies and Recent Accounting Pronouncements.
Overview
Alzamend NeuroTM is a company focused on the facilitation of bringing technologies to market which help with the treatment, prevention or cure of Alzheimer’s Disease.
On May 1, 2016, we obtained a royalty-bearing, exclusive worldwide license from the University, to a mutant-peptide immunotherapy that is designed to be used both as a vaccine and prophylactic against Alzheimer’s. This peptide, known as CAO22W, has transitioned from early stage development to an extensive program of preclinical study and evaluation with an anticipated completion date at the end of May 2019. CAO22W will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it can provide us with any revenue. We plan to file an IND with the FDA with respect to CAO22W in the second quarter of 2019 and prepare to conduct a Phase 1 Clinical Trial at Emory University Alzheimer’s Disease Research Center starting in the latter half of 2019. Upon FDA approval of the IND, we plan to work with Dr. Chuanhai Cao, who is associated with the College of Chemistry, a neuroscientist at the University of South Florida Health Byrd Alzheimer’s Institute and the inventor of CAO22W, and Dr. Cao’s medical and biotech research team to launch our Phase 1 Clinical Trial with up to 30 human patients.
Recent Developments
On July 2, 2018, we obtained two royalty-bearing, exclusive worldwide licenses from the University, to a therapy known as LiProSalTM to mitigate extreme agitation and forestall other deterioration as displayed by patients with up to moderate AD. LiProSalTM is an ionic cocrystal of lithium for the treatment of Alzheimer’s and possibly other neurodegenerative diseases. Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of AD. Unlike traditional medications, which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, it exerts neuroprotective effects, in part, by increasing the brain-derived neurotrophic factor leading to restoration of learning and memory based on the results with some of the animal subjects during the preclinical studies. Another neuroprotective mechanism of lithium is attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Alzamend is continuing to work with two of the inventors of this therapeutic, both at the USF Morsani College of Medicine; Jun Tan, PhD, MD, Professor at the College of Medicine Neurosurgery, Endowed Chair, College of Medicine Psychiatry and Behavioral Neurosciences and the Silver Endowed Chair in Developmental Neurobiology, Department of Psychiatry & Behavioral Medicine and Roland D. Shytle, PhD, Associate Professor, Center of Excellence for Aging & Brain Repair. Dr. Tan and Dr. Shytle with Adam J. Smith, PhD., MSBE, Michael Zaworotko, PhD., and Naga Duggirala, PhD., all formerly with the University of South Florida, have designed, synthesized and characterized this ionic cocrystal of lithium, which Alzamend has named LiProSal. LiProSalTM has been shown to exhibit improved pharmacokinetics compared to current FDA-approved lithium-based drugs as well as bioactive in many in vitro models of Alzheimer’s disease. LiProSalTM may prove to be a major improvement over current lithium-based treatments and may also represent a means of treating AD.
On May 29, 2018, the Company implemented a 1-for-4 Reverse Stock Split of its Common Stock. The Reverse Stock Split became effective on December 13, 2018. As a result of the Reverse Stock Split, every four (4) shares of the Company’s pre-Reverse Stock Split Common Stock were combined and reclassified into one share of the Company’s Common Stock. The number of shares of Common Stock subject to outstanding options and warrants were also reduced by a factor of four as of December 13, 2018. All historical share and per-share amounts reflected throughout the consolidated financial statements and other financial information in this filing have been adjusted to reflect the Reverse Stock Split. The par value per share of the Company’s Common Stock was not affected by the Reverse Stock Split.
| 39 |
RESULTS OF OPERATIONS FOR THE YEARS ENDED APRIL 30, 2018 AND 2017
The following table summarizes the results of our operations for the years ended April 30, 2018 and 2017.
ALZAMEND NEURO, INC.
Condensed Statements of Operations
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| OPERATING EXPENSES | ||||||||
| Research and development | $ | 323,403 | $ | 275,047 | ||||
| General and administrative | 575,027 | 1,273,350 | ||||||
| Total operating expenses | 898,430 | 1,548,397 | ||||||
| Loss from operations | (898,430 | ) | (1,548,397 | ) | ||||
| OTHER EXPENSE, NET | ||||||||
| Interest income - related party | 7,341 | - | ||||||
| Interest expense | (30,259 | ) | (11,390 | ) | ||||
| Interest expense - debt discount | (10,315 | ) | (10,111 | ) | ||||
| Total other expense, net | (33,233 | ) | (21,501 | ) | ||||
| NET LOSS | $ | (931,663 | ) | $ | (1,569,898 | ) | ||
| Basic and diluted net loss per common share | $ | (0.02 | ) | $ | (0.05 | ) | ||
| Basic and diluted weighted average common | 37,889,196 | 28,801,309 | ||||||
| shares outstanding | ||||||||
Revenue
Alzamend Neuro, Inc. is a development stage company that was formed on February 26, 2016. The Company was formed to acquire and commercialize patented intellectual property and know how to prevent, treat and cure the crippling and deadly disease, Alzheimer’s. At that time, Alzamend had only one product candidate, CAO22W, which is a mutant-peptide immunotherapy that is designed to be used both as a vaccine and prophylactic against Alzheimer’s. CAO22W is beginning to complete its preclinical stage of development and will require extensive clinical study, review and evaluation, regulatory review and approval, significant marketing efforts and substantial investment before CAO22W and any successors could provide us with any revenue. The Company did not generate any revenues during the years ended April 30, 2018 and 2017, and we do not anticipate that we will generate revenue for the foreseeable future.
| 40 |
General and administrative expenses
General and administrative expenses for the years ending April 30, 2018 and 2017 were $575,027 and $1,273,350, respectively. As reflected in the table below, general and administrative expenses primarily consisted of the following expense categories: management services, professional fees, and advertising and promotion. The remaining general and administrative expenses of $55,298 and $109,636, respectively, primarily consisted of payments for transfer agent fees, travel, and other office expenses, none of which is significant individually.
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Management Services | $ | 240,000 | $ | 420,000 | ||||
| Professional fees | 249,820 | 412,875 | ||||||
| Advertising and promotion | 29,909 | 330,839 | ||||||
| Other general and administrative expenses | 55,298 | 109,636 | ||||||
| Total general and administrative expenses | $ | 575,027 | $ | 1,273,350 | ||||
Management services
The Company had no full-time employees during the years ended April 30, 2018 and 2017. The services of the two officers and Executive Chairman of the Company are provided pursuant to the terms of an MSA entered into with Avalanche, a related party, on May 1, 2016. Avalanche provides management, consulting and financial services to the Company. Such services include advice and assistance concerning any and all aspects of operations, planning and financing of Alzamend and conducting relations with accountants, attorney, financial advisors and other professionals. The term of the MSA, as amended, is for the period May 1, 2016 to December 31, 2017 and was extended by written agreement. The Company initially paid $40,000 per month for these services and, beginning February 2017, began paying $20,000 per month. During the years ended April 30, 2018 and 2017, the Company recognized $240,000 and $420,000, respectively, in management fees. At April 30, 2018 and April 30, 2017, $3,000 and $180,000, respectively, was included within related party payable on the Company’s balance sheet. The MSA was terminated as of December 31, 2018.
Professional fees
The second largest component of our general and administrative expenses is professional fees. During the years ended April 30, 2018 and 2017, the Company reported professional fees of $249,820 and $412,875, respectively, which are principally comprised of the following items:
Year Ended April 30, 2018
| · | During the year ended April 30, 2018, the Company incurred $82,202 in audit fees. |
| · | During January 2017, the Company entered into consulting agreements with two consultants. Both consulting agreements had a term of one year and provided for aggregate compensation of $50,000. The consulting services included assistance in evaluating strategic opportunities, business advice and services related to sales, marketing, investor relation and other incidental services on behalf of the Company. During the year ended April 30, 2018, the Company recorded an expense of $35,617 as a result of these two consulting agreements. |
| · | The Company entered into a five year consulting agreement with Spartan pursuant to which Spartan has agreed to provide consulting services with respect to general corporate matters, including, but not limited to, advice and input with respect to raising capital, potential merger and acquisition transactions, identifying suitable personnel for management, developing corporate structure and finance strategies, assisting the Company with strategic introductions, assisting management with enhancing corporate and shareholder value and introducing the Company to potential investors. In December 2017, since the maximum amount was raised in a prior private placement, the Company paid to Spartan a consulting fee of $1,400,000 for the services to be rendered over the sixty (60) month term of this consulting agreement. During the year ended April 30, 2018, the Company recorded an expense of $93,333 as a result of this consulting agreement. |
| 41 |
Year Ended April 30, 2017
| · | During the year ended April 30, 2017, as a result of a consulting agreement with Hallmark Investments, LLC (“Hallmark”), the Company incurred $120,000 in professional fees. Effective May 1, 2016, the Company entered into a one-year Consulting Agreement with Hallmark. The terms of the Consulting Agreement provided for $120,000 in payments to Hallmark for strategic advisory services related to the operations of the Company. |
| · | During the year ended April 30, 2017, the Company incurred $72,985 for information technology and website development services from two vendors. |
| · | During the year ended April 30, 2017, the Company incurred $76,625 in audit and legal fees related to the Company’s annual audit of its financial statements for the year ended April 30, 2016, audit related fees attributed to the filing of the Company’s Prior Offering Statement and legal fees resulting from work on the Company’s various regulatory and financing matters. |
| · | The Company incurred $57,447 in stock-based compensation expense from the issuance of 337,500 shares of Common Stock to consultants for sales, marketing, investor relation and other incidental services. |
| · | The remaining amounts attributed to professional fees incurred by the Company during the year ended April 30, 2017 are attributed to various types of professional fees, such as FDA consulting services, none of which is significant individually. |
Advertising and promotion
During the years ended April 30, 2018 and 2017, the Company incurred $29,909 and $330,839, respectively, in advertising and promotion related expenses. The majority of these expenditures were related to direct advertising of the Company’s Offering Statement on Google and Facebook, the development of a social media strategy, costs associated with an inbound call center and the development of videos to convey the Company’s mission.
Research and development expenses
Research and development expenses for the years ending April 30, 2018 and 2017 were $323,403 and $275,047, respectively. As reflected in the table below, research and development expenses primarily consisted of professional fees as well as licenses and fees.
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Professional fees | $ | 101,277 | $ | 5,882 | ||||
| Licenses and fees | 219,163 | 269,165 | ||||||
| Other research and development expenses | 2,963 | - | ||||||
| Total research and development expenses | $ | 323,403 | $ | 275,047 | ||||
Professional fees
The largest component of our research and development expenses is professional fees. During the years ended April 30, 2018 and 2017, the Company reported professional fees of $101,277 and $5,882, respectively, which are principally comprised of professional fees incurred by the Company during the year ended April 30, 2018 are attributed to various types of scientific services, including FDA consulting services.
| 42 |
Licenses and fees
There are certain initial license fees and milestone payments required to be paid to the University of South Florida and the USF Health Byrd Alzheimer’s Institute, a multi-disciplinary center at the University of South Florida, for the licenses of the Technologies, pursuant to the terms of the License Agreement with the Licensor and a direct support organization of the University.
The License Agreement for CAO22W requires, in addition to royalty payments of 4% on net sales of products developed from the licensed technology, the Company to pay a license fee of $100,000 on June 25, 2016 and December 31, 2016. As an additional licensing fee, Licensor is entitled to receive that number of shares of the Company’s Common Stock equal to five percent (5%) of the total number of issued and outstanding shares outstanding on May 1, 2016, subject to additional issuances until such time as the Company has received a total of $5 million in cash in exchange for the Company’s equity securities. Additionally, the Company is required to pay milestone payments to Licensor for the license of the technology.
During the years ended April 30, 2018 and 2017, the Company incurred $218,416 and $56,962, respectively, in non-cash charges from issuances of Common Stock to the Licensor.
Other (expense) income, net
During the year ended April 30, 2018, the Company reported other expense of $33,233. Other expense includes interest expense of $30,259 and amortization of original issue discounts on notes payable and notes payable, related parties of $10,315, partially offset by $7,341 of interest income, related parties.
During the year ended April 30, 2017, the Company reported other expense of $21,501. Other expense includes interest expense of $11,390 and amortization of original issue discounts on notes payable and notes payable to related parties of $10,111.
At April 30, 2018, the Company had no outstanding borrowings. At April 30, 2017, the aggregate outstanding balance on the Company’s borrowings was $325,211 consisting of notes payable of $71,382 and notes payable, related parties of $253,829.
Current and deferred income taxes
The Company has made the decision to fully reserve its net deferred tax assets. As a result of this decision, the Company did not record an income tax benefit during the year ended April 30, 2018 and 2017.
The ultimate realization of deferred tax assets is dependent upon the existence, or generation, of taxable income in the periods when those temporary differences and net operating loss carryovers are deductible. Management considers the scheduled reversal of deferred tax liabilities, taxes paid in carryover years, projected future taxable income, available tax planning strategies, and other factors in making this assessment. Based on available evidence, management believes it is less likely than not that all of the deferred tax assets will be realized. Accordingly, the Company has established a 100% valuation allowance.
LIQUIDITY AND CAPITAL RESOURCES
The accompanying consolidated financial statements have been prepared on the basis that the Company will continue as a going concern. As of April 30, 2018, the Company had cash of $545,001 and an accumulated deficit of $2,513,137. The Company has incurred recurring losses and reported losses for the year ended April 30, 2018 totaled $931,663. In the past, the Company has financed its operations principally through issuances of promissory notes and equity securities. During the year ended April 30, 2018, the Company continued to successfully obtain additional equity and debt financing.
| 43 |
The Company expects to continue to incur losses for the foreseeable future and needs to raise additional capital until it is able to generate revenues from operations sufficient to fund its development and commercial operations. Based on our current business plan, we believe that our cash and cash equivalents at April 30, 2018, are not sufficient to meet our anticipated cash requirements during the twelve-month period subsequent to the issuance of the financial statements included in this Annual Report on Form 1-K. Management believes that the Company has access to capital resources through potential public or private issuance of debt or equity securities. However, the Company cannot be certain that additional funding will be available on acceptable terms, or at all, in which case it may have to significantly delay, scale back or discontinue the development and/or commercialization of its product. The Company may also be required to (a) seek collaborators for its product at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; or (b) relinquish or otherwise dispose of rights to technology or its product that the Company would otherwise seek to deploy or commercialize. These matters raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might become necessary should the Company be unable to continue as a going concern.
On April 10, 2018, Avalanche issued a promissory note to the Company (the “AVLP Note”) to evidence the Company’s loan of up to $995,500 for a period ending on April 30, 2019, subject to the terms and conditions stated in the AVLP Note. The AVLP Note accrues interest at 10% per annum and includes a 10% original issue discount. At April 30, 2018, the Company has provided loans to Avalanche in the principal amount of $800,000, of which $400,000 was repaid, resulting in net loans to Avalanche of $400,000 and an original issue discount of $40,000.
During January 2017, the Company entered into a promissory note and received net proceeds of $65,000. As consideration for this loan, the Company issued a promissory note in the aggregate principal amount of $75,000, which included an OID and fees of $10,000. This loan was repaid during the year ended April 30, 2018.
At April 30, 2017, the outstanding balance on short term borrowings from Avalanche, a related party, was $180,100. During the year ended April 30, 2018, Avalanche provided an additional $123,500 in short term financing. This short-term obligation was non-interest bearing, due upon demand and repaid during the year ended April 30, 2018.
During January 2017, the Company entered a promissory note and received net proceeds of $70,000 from Gary Gottlieb, a related party. As consideration for this loan, the Company issued a promissory note in the aggregate principal amount of $80,000, which included an OID of $10,000. This promissory note accrued interest at 15% per year and during the years ended April 30, 2018 and 2017. The Company repaid this loan during the year ended April 30, 2018.
On October 1, 2017, the Company entered into a promissory note in the principal amount of $47,520 with DPW Holdings, Inc., a related party (“DPW”). The promissory note included an OID of $3,520 resulting in net proceeds to the Company of $44,000, yielded 8% simple interest on the principal amount, and was due on August 16, 2018. Prior to April 30, 2018, the Company repaid the outstanding balance to DPW.
Between December 13, 2017 and December 29, 2017, the Company entered into subscription agreements for the purchase of 311.75 units at $10,000 for each unit purchased. Each unit consisted of 10,000 shares of Common Stock. In aggregate, the 311.75 units represented 3,117,500 shares of Common Stock for an aggregate purchase price of $3,117,500, or $1.00 per share, pursuant to the terms of a Private Placement Memorandum dated August 17, 2017 (the “PPM”). In conjunction with this PPM, and inclusive of the sale of 107.7 units that occurred on October 19, 2017, the Company incurred $419,450 in placement fees and $57,900 in legal and filing fees, resulting in net proceeds to the Company of $3,717,150.
CONTRACTUAL OBLIGATIONS
On May 1, 2016, the Company entered into the License Agreement with the Licensor pursuant to which the Licensor granted the Company a royalty bearing, exclusive worldwide license, limited to the field of Alzheimer’s Immunotherapy and Diagnostics, under United States Patent No. 8,188,046, entitled “Amyloid Beta Peptides and Methods of Use”, filed April 7, 2009 and granted May 29, 2012.
| 44 |
In addition to royalty payments of 4% on net sales of products developed from the licensed technology, the Company was required to pay a license fee of $100,000 on June 25, 2016 and December 31, 2016. As an additional licensing fee, the Licensor is entitled to receive that number of shares of the Company’s Common Stock equal to five percent (5%) of the sum of the total number of issued and outstanding shares plus any securities that are convertible into or exercisable or exchangeable for shares of Common Stock, subject to adjustment for additional issuances until such time as the Company has received a total of $5 million in cash in exchange for the Company’s equity securities. Additionally, the Company is required to pay milestone payments on the due dates to Licensor for the license of the technology, as follows:
| Payment | Due Date | Event | ||||
| $ | 50,000 | September 30, 2019 | Upon IDN application filing | |||
| $ | 50,000 | 12 months from IND application filing date | Upon first dosing of patient in first Phase I Clinical Trial | |||
| $ | 175,000 | 12 months from first patient dosed in Phase I | Upon Completion of first Phase I Clinical Trial | |||
| $ | 500,000 | 24 months from completion of first Phase I Trial | Upon Completion of first Phase II Clinical Trial | |||
| $ | 1,000,000 | 12 months from completion of the first Phase II Clinical Trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA BLA Approval | |||
None of these milestones was met as of the date of this Annual Report. If we fail to meet a milestone by its specified date, the Licensor may terminate the License Agreement.
Licensor was also granted a preemptive right to acquire such shares or other equity securities that may be issued from time to time by the Company while Licensor remains the owner of any equity securities of the Company. Further, if the Company issues equity securities at a price per share that is less than the price paid by purchasers in a transaction for aggregate consideration of at least $5,000,000 (the “Investment Price”), then the number of shares owned by Licensee shall be increased upon such issuance. The amount of the increase shall be determined by multiplying the number of shares then owned by Licensor by a fraction; the numerator of which shall be equal to the number of shares of Common Stock outstanding immediately after the issuance of additional shares of commons stock, and the denominator of which shall be equal to the sum of (i) the number of shares of Common Stock outstanding immediately prior to the issuance of additional shares of Common Stock plus (ii) the number of shares of Common Stock which the aggregate consideration for the total number of additional shares of Common Stock so issued would purchase at the Investment Price.
There are certain license fees and milestone payments required to be paid for the licensing of the LiProSalTM technology, pursuant to the terms of the Standard Exclusive License Agreements with Sublicensing Terms, both dated June 21, 2018 (the “LiProSalTM License Agreements”) with the Licensor and the University. In addition, a royalty payment of 3% is required pursuant to License #18110 while License #1811 requires a royalty payment of 1.5% on net sales of products developed from the licensed technology. For the two LiProSalTM licenses, in the aggregate, the Company is required to pay initial license fees of $50,000 no later than July 31, 2018 and $150,000 no later than October 31, 2018, although the Company is in discussions with the University to defer the payment due date of the initial license fees. As an additional licensing fee, the Licensor is entitled to receive that number of shares of Common Stock equal to three percent (3%) of the sum of the total number of issued and outstanding shares. Additionally, the Company is required to pay milestone payments on the due dates to Licensor for the license of the technology, as follows:
| Payment | Due Date | Event | ||||
| $ | 50,000 | November 1, 2019 | Pre-IND meeting | |||
| $ | 65,000 | 6 months from IND filing date | IND application filing | |||
| $ | 190,000 | 12 months from IND filing date | Upon first dosing of patient in a clinical trial | |||
| $ | 500,000 | 12 months from first patient dosing | Upon Completion of first clinical trial | |||
| $ | 1,250,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III Clinical Trial | |||
| $ | 10,000,000 | 8 years from the Effective Date of the Agreement | Upon FDA Approval | |||
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, changes in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources that is material to investors.
| 45 |
Item 3. Directors, Executive Officers and Corporate Governance.
The following table sets forth information regarding our executive officers, directors and significant employees, including their ages as of the date of this Annual Report:
| Name | Age | Position | ||
| Stephan Jackman | 44 | Chief Executive Officer | ||
| Kenneth S. Cragun | 58 | Chief Financial Officer | ||
| Milton C. Ault, III | 49 | Executive Chairman & Director | ||
| Philip E. Mansour | 50 | Director | ||
| William B. Horne | 51 | Director |
Directors serve until the next annual meeting and until their successors are elected and qualified. Officers are appointed to serve for one year until the meeting of the Board following the annual meeting of stockholders and until their successors have been elected and qualified.
Stephan Jackman
Mr. Jackman has played an intricate role in the development of therapeutic treatments, products and programs from the research stage to market and commercialization. He has demonstrated a dedicated dual focus of creating value for internal and external stakeholders while developing strategic alliances and cross-function teams to meet and exceed goals. Mr. Jackman has held positions of increasing responsibility at Novartis Pharmaceuticals Corporation, L’Oréal USA, SBM Management Services, and Family Intervention Services. Prior to joining Alzamend NeuroTM, Mr. Jackman was the Chief Operating Officer of Ennaid Therapeutics, an emerging biopharmaceutical focusing on cures for mosquito borne infectious diseases, such as Zika and Dengue viruses. Additionally, he has been an independent project and management consultant assisting start-ups, Fortune 500 companies and non-profits with major strategic initiatives. Mr. Jackman holds a Master’s of Science, Management, and a Bachelor’s of Engineering, Mechanical Engineering, from Stevens Institute of Technology.
We believe Mr. Jackman is qualified to serve as our Chief Executive Officer because of his extensive leadership experience and industry knowledge.
Kenneth S. Cragun
Mr. Cragun served as a CFO Partner at Hardesty, LLC, a national executive services firm since October 2016. His assignments at Hardesty included serving as CFO of CorVel Corporation, a $1.1 billion market cap publicly traded company (NASDAQ: CRVL) and a nationwide leader in technology driven, healthcare-related, risk management programs and of RISA Tech, Inc. a private structural design and optimization software company. Mr. Cragun was also CFO of two NASDAQ-listed companies, Local Corporation, from April 2009 to September 2016, which operated Local.com, a U.S. top 100 website, and Modtech Holdings, Inc., from June 2006 to March 2009, a supplier of modular buildings. Prior thereto, he had financial leadership roles with increasing responsibilities at MIVA, Inc., ImproveNet, Inc., NetCharge Inc., C-Cube Microsystems, Inc, and 3-Com Corporation. Mr. Cragun is currently the Chief Accounting Officer of DPW Holdings, Inc. and on the Board of Directors and Chairman of the Audit Committee of Verb Technology Company, Inc. (OTC: FUSZD). Mr. Cragun began his professional career at Deloitte. Mr. Cragun holds a Bachelor of Science degree in accounting from Colorado State University-Pueblo.
Mr. Cragun’s industry experience is vast with extensive experience in fast-growth environments and building teams in more than 20 countries. Mr. Cragun has led multiple financing transactions, including IPOs, PIPEs, convertible debt, term loans, and lines of credit. For these reasons, we believe that he is well qualified to service as the Company’s CFO.
Philip E. Mansour
Since October 2008, Mr. Mansour has been the full-time principal of PMC Solutions, LLC, specializing in consulting to companies on issues of operational management, strategic planning, marketing business development and disruptive technology. Additionally, Mr. Mansour has provided executive coaching services. Mr. Mansour has served as a director and as the President and Chief Executive Officer of Avalanche since May 2014. Mr. Mansour worked as the Chief Operational Officer with the RXtra Solutions organization. The organization was a privately-owned set of health care development companies which had footprints in the compounding pharmacy, diagnostics, medical equipment, chemical distribution and wellness provider spaces. Vice President, corporate development for Conceivex, Inc., a private company focused on At-Home Infertility treatment. His prior experience includes leading the research and development for some prominent educational technology companies for more than 2 decades and leading multi-million-dollar government grants with leading universities. His entrepreneurial and significant corporate experience is expected to benefit the Company. We believe that Mr. Mansour’s business background demonstrates he has the qualifications to serve as one of our directors.
| 46 |
Milton C. Ault, III
Mr. Ault founded Alzamend Neuro, Inc., a biotechnology firm dedicated to finding the treatment, prevention and cure for Alzheimer’s Disease on February 25, 2016 and has served as its Executive Chairman since November 2, 2018 and as its Chairman since inception.
Mr. Ault is a seasoned business professional and entrepreneur who has spent more than twenty-seven years identifying value in various financial markets including equities, fixed income, commodities, and real estate. On March 16, 2017, Mr. Ault was appointed Executive Chairman of the Board for DPW Holdings, Inc. (“DPW”), formerly known as Digital Power Corporation (NYSE: DPW) and on December 28, 2017, Mr. Ault was appointed Chief Executive Officer of DPW. Mr. Ault has served as Chairman of Ault & Company, a holding company since December 2015, and as Chairman of Avalanche, a publicly traded company, since September 2014. Since January 2011, Mr. Ault has been the Vice President of Business Development for MCKEA Holdings, LLC, a family office. Mr. Ault has consulted for a few publicly traded and privately held companies, which range from development stage to seasoned businesses, providing each of them the benefit of his diversified experience.
We believe that Mr. Ault’s business background demonstrates he has the qualifications to serve as one of our directors and as Chairman.
William B. Horne.
Mr. Horne has served as director for Alzamend NeuroTM since June 01, 2016. Mr. Horne served as the Chief Financial Officer from June 2016 through December 2018. Mr. Horne is a member of the board of directors of DPW since October 2016. On January 25, 2018, Mr. Horne was appointed as DPW’s Chief Financial Officer. Mr. Horne is a director of and Chief Financial Officer of Avalanche, a publicly traded company. He has served as the Chief Financial Officer of Targeted Medical Pharma, Inc. (OTCBB: TRGM) since August 2013.
Mr. Horne previously held the position of Chief Financial Officer in various companies in the healthcare and high-tech field, including OptimisCorp, from January 2008 to May 2013, a privately held, diversified healthcare technology company located in Los Angeles, California.
Mr. Horne served as the Chief Financial Officer of Patient Safety Technologies, Inc. (OTCBB: PSTX), a medical device company located in Irvine, California, from June 2005 to October 2008 and as the interim Chief Executive Officer from January 2007 to April 2008. In his dual role at Patient Safety Technologies, Mr. Horne was directly responsible for structuring the divestiture of non-core assets, capital financings and debt restructuring.
Mr. Horne has also held supervisory positions at Price Waterhouse, LLP and has a Bachelor of Arts Magna Cum Laude in Accounting from Seattle University.
We believe that Mr. Horne's extensive financial and accounting experience in diversified industries and with companies involving complex transactions give him the qualifications and skills to serve as one of our directors.
Board Leadership Structure and Risk Oversight
The Board oversees our business and considers the risks associated with our business strategy and decisions. The Board currently implements its risk oversight function as a whole. On November 2, 2018, the Board adopted a charter that establishes an Audit Committee and a Nomination & Governance Committee. Each of the Board committees will provide risk oversight in respect of its areas of concentration and reports material risks to the board for further consideration.
Term of Office
Directors serve until the next annual meeting and until their successors are elected and qualified. Officers are appointed to serve for one year until the meeting of the Board following the annual meeting of stockholders and until their successors have been elected and qualified.
| 47 |
Director Independence
We use the definition of “independence” of The NASDAQ Stock Market to make this determination. NASDAQ Listing Rule 5605(a)(2) provides that an “independent director” is a person other than an officer or employee of the company or any other individual having a relationship which, in the opinion of the Company’s Board, would interfere with the exercise of independent judgment in carrying out the responsibilities of a director. The NASDAQ listing rules provide that a director cannot be considered independent if:
| · | the director is, or at any time during the past three years was, an employee of the company; |
| · | the director or a family member of the director accepted any compensation from the company in excess of $120,000 during any period of 12 consecutive months within the three years preceding the independence determination (subject to certain exemptions, including, among other things, compensation for board or board committee service); |
| · | the director or a family member of the director is a partner in, controlling stockholder of, or an executive officer of an entity to which the company made, or from which the company received, payments in the current or any of the past three fiscal years that exceed 5% of the recipient’s consolidated gross revenue for that year or $200,000, whichever is greater (subject to certain exemptions); |
| · | the director or a family member of the director is employed as an executive officer of an entity where, at any time during the past three years, any of the executive officers of the company served on the compensation committee of such other entity; or |
| · | the director or a family member of the director is a current partner of the company’s outside auditor, or at any time during the past three years was a partner or employee of the company’s outside auditor, and who worked on the company’s audit. |
Under such definitions, we have no independent directors. However, our Common Stock is not currently quoted or listed on any national exchange or interdealer quotation system with a requirement that a majority of our Board be independent and, therefore, the Company is not subject to any director independence requirements.
Family Relationships
There are no family relationships among any of our officers or directors.
Involvement in Certain Legal Proceedings
Except as disclosed below, to our knowledge, none of our current directors or executive officers has, during the past ten years:
| · | been convicted in a criminal proceeding or been subject to a pending criminal proceeding (excluding traffic violations and other minor offenses); |
| · | had any bankruptcy petition filed by or against the business or property of the person, or of any partnership, corporation or business association of which he was a general partner or executive officer, either at the time of the bankruptcy filing or within two years prior to that time; |
| · | been subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction or federal or state authority, permanently or temporarily enjoining, barring, suspending or otherwise limiting, his involvement in any type of business, securities, futures, commodities, investment, banking, savings and loan, or insurance activities, or to be associated with persons engaged in any such activity; |
| · | been found by a court of competent jurisdiction in a civil action or by the SEC or the Commodity Futures Trading Commission to have violated a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated; |
| 48 |
| · | been the subject of, or a party to, any federal or state judicial or administrative order, judgment, decree, or finding, not subsequently reversed, suspended or vacated (not including any settlement of a civil proceeding among private litigants), relating to an alleged violation of any federal or state securities or commodities law or regulation, any law or regulation respecting financial institutions or insurance companies including, but not limited to, a temporary or permanent injunction, order of disgorgement or restitution, civil money penalty or temporary or permanent cease-and-desist order, or removal or prohibition order, or any law or regulation prohibiting mail or wire fraud or fraud in connection with any business entity; or |
| · | been the subject of, or a party to, any sanction or order, not subsequently reversed, suspended or vacated, of any self-regulatory organization (as defined in Section 3(a)(26) of the Exchange Act), any registered entity (as defined in Section 1(a)(29) of the Commodity Exchange Act), or any equivalent exchange, association, entity or organization that has disciplinary authority over its members or persons associated with a member. |
| 1. | Mr. Ault held series 7, 24, and 63 licenses and managed four domestic hedge funds and one bond fund from 1998 through 2008. On April 26, 2012, as a result of an investigation by FINRA involving activities during 2008, Mr. Ault agreed to a settlement with FINRA in which he did not admit to any liability or violation of any laws or regulatory rules and that included restitution and a suspension from association with a FINRA member firm for a period of 2 years. As part of that settlement, Mr. Ault agreed that he would make restitution to certain investors. Mr. Ault did not within the prescribed time period make a restitution payment to certain of the investors as he was unable to locate all of them, nor did he forward the undistributed restitution in the state where the investor was known to have resided, as directed by FINRA. |
| 2. | Mr. Ault was CEO, President and Chairman of Zealous Holdings, Inc. that filed for bankruptcy protection under Chapter 11 of Title 11 of the United States Code (the “Bankruptcy Code”) on February 20, 2009, in the U.S. Bankruptcy Court, Central District of California. This Chapter 11 filing was subsequently converted to a Chapter 7 filing by order of the Bankruptcy Court. Zealous Holdings, Inc. was not an entity that was entitled to a discharge under the bankruptcy code. As such Zealous Holdings, Inc. did not receive a discharge. Ultimately, Zealous Holdings, Inc. ceased doing business and was permanently closed. |
| 3. | Mr. Ault filed for bankruptcy protection under the Bankruptcy Code on December 8, 2009, in the U.S. Bankruptcy Court, Central District of California. This Chapter 13 filing was subsequently converted to a Chapter 7 filing by order of the Bankruptcy Court and months later, the petition being withdrawn and dismissed without prejudice. |
| 4. | Mr. Cragun served as Chief Financial Officer of Local Corporation (April 2009 to September 2016), formerly based in Irvine, California, and, in June 2015, Local Corporation filed a voluntary petition in the United States Bankruptcy Court for the Central District of California seeking relief under the provisions of Chapter 11 the Bankruptcy Code. |
| 5. | Mr. Cragun served as Chief Financial Officer of Modtech Holdings, Inc. (June 2006 to March 2009), formerly based in Perris, California and, in October 2008, Modtech Holdings, Inc. filed a voluntary petition in the United States Bankruptcy Court for the Central District of California seeking relief under the provisions of Chapter 11 of the Bankruptcy Code. |
Except as set forth in our discussion below in “Certain Relationships and Related Transactions,” none of our directors or executive officers has been involved in any transactions with us or any of our directors, executive officers, affiliates or associates which are required to be disclosed pursuant to the rules and regulations of the SEC.
We are not currently a party to any legal proceedings, the adverse outcome of which, individually or in the aggregate, we believe will have a material adverse effect on our business, financial condition or operating results.
Code of Business Conduct and Ethics
Our Board has adopted a written code of business conduct and ethics, revised effective May 29, 2018, that applies to our directors, officers and employees, including our principal executive officer, principal financial officer and principal accounting officer or controller, or persons performing similar functions. We have posted on our website a current copy of the code and all disclosures that are required by law in regard to any amendments to, or waivers from, any provision of the code.
| 49 |
EXECUTIVE COMPENSATION
The following table represents information, as of October 31, 2018, regarding the total compensation of our executive officers and director of the Company since inception:
| Cash | Other | Total | |||||||
| Compensation | Compensation | Compensation | |||||||
| Name and Principal Position | ($) | ($) (1) | ($) | ||||||
| Philip E. Mansour, President, Chief Executive Officer & Director | — | 600 | 600 | ||||||
| Milton C. Ault, III, Executive Chairman & Director | — | 600 | 600 | ||||||
| William B. Horne, Chief Financial Officer & Director | — | 600 | 600 | ||||||
(1) The values reported in the “Other Compensation” column represents the aggregate grant date fair value, computed in accordance with Accounting Standards Codification (“ASC”) 718 Share Based Payments, of grants of stock options to each of our named executive officers and directors.
No compensation was paid to the officers and directors identified above outside of the MSA with Avalanche. The services of the two officers and Executive Chairman of the Company were provided pursuant to the terms of an MSA entered into with Avalanche, a related party, on May 1, 2016. Pursuant to the terms of the MSA, Avalanche provided management, consulting and financial services to Alzamend. Such services included advice and assistance concerning any and all aspects of operations, planning and financing of Alzamend and conducting relations with accountants, attorney, financial advisors and other professionals. The term of the MSA, as amended, was for the period May 1, 2016 to December 31, 2017 and was extended by written agreement. The Company initially paid $40,000 per month for these services and, beginning February 2017, began paying $20,000 per month. During the years ended April 30, 2018 and 2017, the Company recorded $240,000 and $420,000, respectively, in management fees. At April 30, 2018 and April 30, 2017, $3,000 and $180,000, respectively, was included within related party payable on the Company’s balance sheet.
Item 4. Security Ownership of Management and Certain Securityholders
The following table shows the beneficial ownership of our Common Stock as of February 15, 2019 held by (i) each person known to us to be the beneficial owner of more than five percent (5%) of any class of our shares; (ii) each director; (iii) each executive officer; and (iv) all directors and executive officers as a group. As of February 15, 2019, there were 51,721,119 shares of Common Stock issued and outstanding and 750,000 shares of Series A Preferred Stock issued and outstanding.
Beneficial ownership is determined in accordance with the rules of the Commission, and generally includes voting power and/or investment power with respect to the securities held. Shares of Common Stock subject to options and warrants currently exercisable or which may become exercisable within 60 days of the date of this Annual Report, are deemed outstanding and beneficially owned by the person holding such options or warrants for purposes of computing the number of shares and percentage beneficially owned by such person, but are not deemed outstanding for purposes of computing the percentage beneficially owned by any other person. Except as indicated in the footnotes to this table, the persons or entities named have sole voting and investment power with respect to all shares of Common Stock shown as beneficially owned by them.
The percentages below are based on fully diluted shares of our Common Stock as of October 31, 2018. Unless otherwise indicated, the principal address of each of the persons below is c/o Alzamend Neuro, Inc., 3802 Spectrum Blvd., Suite #112C, Tampa, FL 33612.
| 50 |
| Number of shares of | Percentage of Shares | |||||||
| Common Stock Beneficially Owned | Beneficially Owned | |||||||
| Directors and Officers: | ||||||||
| Phil Mansour (1) | 2,500,000 | 3.09 | % | |||||
| Milton C. Ault, III (2)(3) | 40,000,000 | 49.45 | % | |||||
| William B. Horne (1) | 2,500,000 | 3.09 | % | |||||
| All directors and named executive officers as a group (3 persons) | 22,500,000 | 55.63 | % | |||||
| Greater than 5% Beneficial Owners: | ||||||||
| MCKEA Holdings, LLC (4) | 37,500,000 | 46.36 | % | |||||
| Congregation Chazon Avrohom (5) | 5,902,735 | 7.30 | % | |||||
| University of South Florida (6) | 4,208,920 | 5.20 | % | |||||
| Spartan Capital Securities, LLC (7) | 4,050,000 | 5.01 | % | |||||
(1) Consists of options to purchase 2,500,000 shares of Common Stock.
(2) Includes options to purchase 2,500,000 shares of Common Stock.
(3) Consists of MCKEA Holdings’ 750,000 Series A Preferred Shares that are convertible into 15,000,000 shares of Common Stock but carry the voting power of 37,500,000 shares of Common Stock.
(4) Consists of 750,000 Series A Preferred Shares that are convertible into 15,000,000 shares of Common Stock but carry the voting power of 37,500,000 shares of Common Stock. The control person of MCKEA Holdings, LLC is Kristine L. Ault, Managing Member, the wife of Mr. Ault, the Executive Chairman of the Company.
(5) The control person of the Congregation Chazon Avrohom is Abraham Biderman.
(6) The control person of the University of South Florida is Theile Riordan.
(7) The control person of Spartan Capital Securities, LLC is John Lowry.
Item 5. Interest of Management and Others in Certain Transactions
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Transactions with Related Persons
As of April 30, 2016, the Company had sold 1,000,000 shares of Series A Convertible Preferred Stock to MCKEA Holdings, LLC (“MCKEA”), a related party. The Series A Convertible Preferred Stock is convertible at a rate of 20:1 into shares of the Company’s Common Stock yet carry the voting power on a convertible basis at a rate of 50:1. Kristine L. Ault is the managing member of MCKEA Holdings, LLC and is the wife of Milton C. Ault, III, Chairman of the Company’s Board.
On May 1, 2016, the Company entered into as MSA with Avalanche, a related party. Messrs. Ault, Horne and Mansour are also officers and directors of Avalanche. Further, MCKEA is the majority member of Philou Ventures, LLC, which is the controlling shareholder of Avalanche. Pursuant to the terms of the MSA, Avalanche provided management, consulting and financial services to Alzamend. Such services included advice and assistance concerning any and all aspects of operations, planning and financing of Alzamend and conducting relations with accountants, attorney, financial advisors and other professionals. The term of the MSA, as amended, was for the period May 1, 2016 to December 31, 2017 and was extended by written agreement. The Company initially paid $40,000 per month for these services and, beginning February 2017, began paying $20,000 per month. During the years ended April 30, 2018 and 2017, the Company recognized $240,000 and $420,000, respectively, in management fees. At April 30, 2018 and April 30, 2017, $0 and $180,000, respectively, was included within related party payable on the Company’s balance sheet. The MSA was terminated on December 31, 2018.
| 51 |
On June 28, 2017, MCKEA and Spartan Capital Securities, LLC (“Spartan”) entered into a five-year consulting agreement (the “MCKEA Consulting Agreement”). Pursuant to the MCKEA Consulting Agreement, upon the receipt by the Company of no less than $2,500,000 in gross proceeds from a Private Placement Memorandum dated August 17, 2017, MCKEA transferred to Spartan 5,000,000 shares of Alzamend Common Stock. During the term of the MCKEA Consulting Agreement, Spartan will provide consulting services related to general corporate and other matters related to MCKEA’s investment in the Company such as advice on mergers and acquisition transactions, finance strategies, identification of potential management candidates and other strategic introductions.
The amount due at April 30, 2018 and 2017 to MCKEA and the Company’s officers for reimbursement of expenses paid and incurred by these related parties was $6,636 and $58,373, respectively. The amounts are included within related party payable on the Company’s balance sheet.
There were no amounts due at April 30, 2018 to related parties from short-term loans. The amount due at April 30, 2017 to related parties from short-term loans, inclusive of unamortized original discount of $6,271, was $253,829. The amount is included within notes payable, related parties, on the Company’s balance sheet (See Note 11).
On April 10, 2018, Avalanche, issued the AVLP Note to evidence the Company’s loan of up to $995,500 for the period ending on April 30, 2019, subject to the terms and conditions stated in the AVLP Note. The AVLP Note accrues interest at 10% per annum and includes a 10% original issue discount. During the six months ended October 31, 2018, the Company provided loans to Avalanche in the principal amount of $558,000, of which $386,000 was repaid. As of October 31, 2018, the balance of the loan due to Alzamend is $570,083.
To the best of our knowledge, from inception to our most recent fiscal year end on April 30, 2018, other than as set forth above, there were no material transactions, or series of similar transactions, or any currently proposed transactions, or series of similar transactions, to which we were or are to be a party, in which the amount involved exceeds $120,000, and in which any director or executive officer, or any security holder who is known by us to own of record or beneficially own more than 5% of any class of our Common Stock, or any member of the immediate family of any of the foregoing persons, has an interest (other than compensation to our officers and directors in the ordinary course of business).
Item 6. Other Information
None.
Item 7. Financial Statements
The financial statements required by this Item 7 are included in this Annual Report on the following page.
| 52 |
INDEX TO FINANCIAL STATEMENTS
ALZAMEND NEURO, INC.
| Report of Independent Registered Public Accounting Firm | F-2 |
| Balance Sheets as of April 30, 2018 and 2017 | F-4 |
| Statements of Operations for the years ended April 30, 2018 and 2017 | F-5 |
| Statements of Cash Flows for the years ended April 30, 2018 and 2017 | F-6 |
| Statement of Changes in Stockholders’ Equity (Deficit) for the years ended April 30, 2018 and 2017 | F-7 |
| Notes to Financial Statements | F-8 – F-21 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED ACCOUNTING FIRM
To the Board of Directors and Stockholders of Alzamend Neuro, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheet of Alzamend Neuro, Inc. (the “Company”) as of April 30, 2018, and the related statement of operations, changes in stockholders’ equity (deficit) and cash flows for the year then ended and the related notes to the financial statements (collectively, the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at April 30, 2018, and the results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
Going Concern Uncertainty
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has a history of significant recurring losses from operations through April 30, 2018, and does not have sufficient working capital at April 30, 2018 to fund its planned operations during the twelve-month period subsequent to the issuance of these financial statements. This raises substantial doubt about the Company's ability to continue as a going concern. Management's plans in regard to these matters also are described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audit provide a reasonable basis for our opinion.
/s/ SQUAR MILNER LLP
We have served as the Company's auditor since 2019.
San Diego, California
February 21, 2019
| F-2 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
of Alzamend Neuro, Inc.
We have audited the accompanying balance sheet of Alzamend Neuro, Inc. (the “Company”) as of April 30, 2017, and the related statements of operations, changes in stockholders’ deficit and cash flows for the year then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audit included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audit provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Alzamend Neuro, Inc., as of April 30, 2017, and the results of its operations and its cash flows for the year then ended in conformity with accounting principles generally accepted in the United States of America.
/s/ Marcum llp
New York, NY
January 29, 2018, except for the reverse stock split disclosed in Note 1, which is dated February 21, 2019
| F-3 |
ALZAMEND NEURO, INC.
Balance Sheets
| April 30, 2018 | April 30, 2017 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash | $ | 545,001 | $ | 4,976 | ||||
| Note receivable, related party, net | 403,333 | - | ||||||
| Prepaid expenses and other currrent assets | 1,467,685 | 52,317 | ||||||
| TOTAL CURRENT ASSETS | 2,416,019 | 57,293 | ||||||
| Deferred offering cost | - | 75,000 | ||||||
| TOTAL LONG TERM ASSETS | - | 75,000 | ||||||
| TOTAL ASSETS | $ | 2,416,019 | $ | 132,293 | ||||
| LIABILITIES AND STOCKHOLDERS’ DEFICIT | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable and accrued liabilities | $ | 90,088 | $ | 125,965 | ||||
| Related party payable | 6,636 | 266,047 | ||||||
| Notes payable, net of discount of $0 and $3,618, respectively | - | 71,382 | ||||||
| Notes payable, related party, net of discount of $5,857 and $6,271, respectively | - | 253,829 | ||||||
| TOTAL CURRENT LIABILITIES | 96,724 | 717,223 | ||||||
| COMMITMENTS AND CONTINGENCIES | ||||||||
| STOCKHOLDERS’ EQUITY (DEFICIT) | ||||||||
| Convertible Preferred stock, $0.0001 par value: 10,000,000 shares authorized; | ||||||||
| Series A Preferred Stock, $0.0001 stated value per share, 1,360,000 shares designated; | ||||||||
| 750,000 and 1,360,000 shares issued and outstanding as of April 30, 2018 and 2017, respectively | 75 | 136 | ||||||
| Common stock, $0.0001 par value: 300,000,000 shares authorized; | ||||||||
| 49,493,196 and 32,874,342 shares issued and outstanding as of April 30, 2018 | ||||||||
| and April 30, 2017, respectively | 4,949 | 3,287 | ||||||
| Additional paid-in capital | 4,827,408 | 993,121 | ||||||
| Accumulated deficit | (2,513,137 | ) | (1,581,474 | ) | ||||
| TOTAL STOCKHOLDERS’ EQUITY (DEFICIT) | 2,319,295 | (584,930 | ) | |||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | $ | 2,416,019 | $ | 132,293 | ||||
The accompanying notes are an integral part of these financial statements.
| F-4 |
ALZAMEND NEURO, INC.
Statements of Operations
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| OPERATING EXPENSES | ||||||||
| Research and development | $ | 323,403 | $ | 275,047 | ||||
| General and administrative | 575,027 | 1,273,350 | ||||||
| Total operating expenses | 898,430 | 1,548,397 | ||||||
| Loss from operations | (898,430 | ) | (1,548,397 | ) | ||||
| OTHER EXPENSE, NET | ||||||||
| Interest income - related party | 7,341 | - | ||||||
| Interest expense | (30,259 | ) | (11,390 | ) | ||||
| Interest expense - debt discount | (10,315 | ) | (10,111 | ) | ||||
| Total other expense, net | (33,233 | ) | (21,501 | ) | ||||
| NET LOSS | $ | (931,663 | ) | $ | (1,569,898 | ) | ||
| Basic and diluted net loss per common share | $ | (0.02 | ) | $ | (0.05 | ) | ||
| Basic and diluted weighted average common shares outstanding | 37,889,196 | 28,801,309 | ||||||
The accompanying notes are an integral part of these financial statements.
| F-5 |
ALZAMEND NEURO, INC.
Statements of Cash Flows
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | (931,663 | ) | $ | (1,569,898 | ) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Interest expense -- debt discount | 10,315 | 10,111 | ||||||
| Legal expense -- debt discount | 1,809 | - | ||||||
| Accretion of original issue discount on notes receivable – related party | (3,333 | ) | - | |||||
| Stock-based compensation for license fees | 218,417 | 56,962 | ||||||
| Stock-based compensation to employees, directors and consultants | - | 57,447 | ||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | (1,415,368 | ) | (49,679 | ) | ||||
| Deferred offering costs | 75,000 | - | ||||||
| Accounts payable | (6,877 | ) | 78,363 | |||||
| Net cash used in operating activities | (2,051,700 | ) | (1,416,694 | ) | ||||
| Cash flows from investing activities: | ||||||||
| Loans to related party | (840,000 | ) | - | |||||
| Proceeds from loans to related party | 440,000 | - | ||||||
| Net cash used in investing activities | (400,000 | ) | - | |||||
| Cash flows from financing activities: | ||||||||
| Proceeds from issuance of common stock | 3,615,236 | 866,620 | ||||||
| Advances from party payable | (259,411 | ) | 262,929 | |||||
| Deferred offering costs | - | (30,000 | ) | |||||
| Proceeds from issuance of preferred stock | - | 5,880 | ||||||
| Proceeds from notes payable | - | 140,000 | ||||||
| Proceeds from notes payable, related party | 138,500 | 250,100 | ||||||
| Payments on notes payable | (75,000 | ) | - | |||||
| Payments on notes payable, related party | (427,600 | ) | (75,000 | ) | ||||
| Net cash provided by financing activities | 2,991,725 | 1,420,529 | ||||||
| Net increase in cash | 540,025 | 3,835 | ||||||
| Cash at beginning of period | 4,976 | 1,141 | ||||||
| Cash at end of period | $ | 545,001 | $ | 4,976 | ||||
| Supplemental disclosures of cash flow information: | ||||||||
| Cash paid during the period for interest | $ | 30,569 | $ | 5,000 | ||||
| Non-cash financing activities: | ||||||||
| Subscription receivable for common stock | $ | - | $ | 1,700 | ||||
| Fair value of warrants issued in connection with promissory note | $ | 2,235 | $ | - | ||||
| Deferred offering costs included in accounts payable | $ | - | $ | 45,000 | ||||
| Issuance of notes payable in payment of accrued expenses | $ | 29,000 | $ | - | ||||
| Issuance of common stock upon conversion of convertible preferred stock | $ | 1,220 | $ | - | ||||
The accompanying notes are an integral part of these financial statements.
| F-6 |
ALZAMEND NEURO, INC.
Statements of Changes in Stockholders’ Equity (Deficit)
Years Ended April 30, 2018 and April 30, 2017
| Receivable | ||||||||||||||||||||||||||||||||
| Series A Convertible | Additional | due from | ||||||||||||||||||||||||||||||
| Preferred Stock | Common Stock | Paid-In | Preferred | Accumulated | ||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Capital | Shareholder | Deficit | Total | |||||||||||||||||||||||||
| BALANCES, April 30, 2016 | 1,000,000 | $ | 100 | 11,600,000 | $ | 1,160 | $ | 13,210 | $ | (3,000 | ) | $ | (11,576 | ) | $ | (106 | ) | |||||||||||||||
| Issuance of common stock | - | - | 17,550,000 | 1,755 | 863,030 | - | - | 864,785 | ||||||||||||||||||||||||
| Issuance of common stock for services | - | - | 337,500 | 34 | 57,413 | - | - | 57,447 | ||||||||||||||||||||||||
| Issuance of common stock for license fees | - | - | 3,386,842 | 338 | 56,624 | - | - | 56,962 | ||||||||||||||||||||||||
| Issuance of preferred stock | 360,000 | 36 | - | 2,844 | 3,000 | 5,880 | ||||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (1,569,898 | ) | (1,569,898 | ) | ||||||||||||||||||||||
| BALANCES, April 30, 2017 | 1,360,000 | 136 | 32,874,342 | 3,287 | 993,121 | - | (1,581,474 | ) | (584,930 | ) | ||||||||||||||||||||||
| Issuance of common stock | - | - | 4,203,887 | 421 | 3,614,815 | - | - | 3,615,236 | ||||||||||||||||||||||||
| Issuance of common stock for conversion | ||||||||||||||||||||||||||||||||
| of Series A preferred stock | (610,000 | ) | (61 | ) | 12,200,000 | 1,220 | (1,159 | ) | - | - | - | |||||||||||||||||||||
| Issuance of common stock for license fees | - | - | 214,967 | 21 | 218,396 | - | - | 218,417 | ||||||||||||||||||||||||
| Fair value of warrants issued in connection | ||||||||||||||||||||||||||||||||
| with promissory note | - | - | - | - | 2,235 | - | - | 2,235 | ||||||||||||||||||||||||
| Net loss | - | - | - | - | - | - | (931,663 | ) | (931,663 | ) | ||||||||||||||||||||||
| BALANCES, April 30, 2018 | 750,000 | $ | 75 | 49,493,196 | $ | 4,949 | $ | 4,827,408 | $ | — | $ | (2,513,137 | ) | $ | 2,319,295 | |||||||||||||||||
The accompanying notes are an integral part of these financial statements.
| F-7 |
ALZAMEND NEURO, INC.
NOTES TO FINANCIAL STATEMENTS
1. DESCRIPTION OF BUSINESS
Alzamend Neuro, Inc. (the “Company” or “Alzamend”), is a specialty pharmaceutical company that was formed on February 26, 2016 to develop and commercialize patented intellectual property to prevent, treat and cure Alzheimer’s. The first patented solution that Alzamend has licensed to move to commercialization is an immunotherapy vaccine peptide that works both as a treatment and vaccine against Alzheimer’s (the “Technology”).
On May 29, 2018, the Company implemented a 1-for-4 Reverse Stock Split of its Common Stock. The Reverse Stock Split became effective on December 13, 2018. As a result of the Reverse Stock Split, every four (4) shares of the Company’s pre-Reverse Stock Split Common Stock were combined and reclassified into one share of the Company’s Common Stock. The number of shares of Common Stock subject to outstanding options and warrants were also reduced by a factor of four as of December 13, 2018. All historical share and per-share amounts reflected throughout the consolidated financial statements and other financial information in this filing have been adjusted to reflect the Reverse Stock Split. The par value per share of the Company’s Common Stock was not affected by the Reverse Stock Split.
The Company is devoting substantially all of its efforts towards research and development of its Technology and raising capital. The Company has not generated any product revenue to date. The Company has financed its operations to date primarily through debt financings and through the sale of its Common Stock. The Company expects to continue to incur net losses in the foreseeable future.
2. LIQUIDITY, GOING CONCERN AND MANAGEMENT’S PLANS
The accompanying consolidated financial statements have been prepared on the basis that the Company will continue as a going concern. As of April 30, 2018, the Company had cash of $545,001 and an accumulated deficit of $2,513,137. The Company has incurred recurring losses for the year ended April 30, 2018 totaling $931,663. In the past, the Company has financed its operations principally through issuances of promissory notes and equity securities. During the year ended April 30, 2018, the Company continued to successfully obtain additional equity and debt financing.
The Company expects to continue to incur losses for the foreseeable future and needs to raise additional capital until it is able to generate revenues from operations sufficient to fund its development and commercial operations. Based on our current business plan, we believe that our cash and cash equivalents at April 30, 2018, are not sufficient to meet our anticipated cash requirements during the twelve-month period subsequent to the issuance of the financial statements included in this Annual Report on Form 1-K. Management believes that the Company has access to capital resources through potential public or private issuance of debt or equity securities. However, the Company cannot be certain that additional funding will be available on acceptable terms, or at all, in which case it may have to significantly delay, scale back or discontinue the development and/or commercialization of its product. The Company may also be required to (a) seek collaborators for its product at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; or (b) relinquish or otherwise dispose of rights to technology or its product that the Company would otherwise seek to deploy or commercialize. These matters raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying financial statements do not include any adjustments that might become necessary should the Company be unable to continue as a going concern.
3. SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”) and pursuant to the rules and regulations of the Securities and Exchange Commission.
| F-8 |
Accounting Estimates
The preparation of financial statements, in conformity with U.S. GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. The Company’s critical accounting policies that involve significant judgment and estimates include share-based compensation and valuation of deferred income taxes. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments with a remaining maturity of three months or less when purchased to be cash equivalents. The recorded carrying amounts of the Company’s cash and cash equivalents approximate their fair value. As of April 30, 2018 and 2017, the Company had no cash equivalents.
Fair Value of Financial Instruments
The Company’s financial instruments are accounts payable, notes payable and notes payable, related party. The recorded values of accounts payable approximate their fair values based on their short-term nature. The recorded values of notes payable and notes payable, related party are recorded at their carrying value, net of any unamortized debt discount, which approximates their fair value based on their short-term nature.
U.S. GAAP defines fair value as the exchange price that would be received for an asset or paid to transfer a liability (an exit price) in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The fair value hierarchy is based on three levels of inputs that may be used to measure fair value, of which the first two are considered observable and the last is considered unobservable:
Level 1: Quoted prices in active markets for identical assets or liabilities.
Level 2: Inputs other than Level 1 that are observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities.
Level 3 assumptions: Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities including liabilities resulting from imbedded derivatives associated with certain warrants to purchase Common Stock.
Income Taxes
The Company determines its income taxes under the asset and liability method. Under the asset and liability approach, deferred income tax assets and liabilities are calculated and recorded based upon the future tax consequences of temporary differences by applying enacted statutory tax rates applicable to future periods for differences between the financial statements carrying amounts and the tax basis of existing assets and liabilities. Generally, deferred income taxes are classified as current or non-current in accordance with the classification of the related asset or liability. Those not related to an asset or a liability are classified as current or non-current depending on the periods in which the temporary differences are expected to reverse. Valuation allowances are provided for significant deferred income tax assets when it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company recognizes tax liabilities by prescribing a minimum probability threshold that a tax position must meet before a financial statement benefit is recognized and also provides guidance on de-recognition, measurement, classification, interest and penalties, accounting in interim periods, disclosure and transition. The minimum threshold is defined as a tax position that is more likely than not to be sustained upon examination by the applicable taxing authority, including resolution of any related appeals or litigation processes, based on the technical merits of the position. The tax benefit to be recognized is measured as the largest amount of benefit that is greater than fifty percent likely of being realized upon ultimate settlement. To the extent that the final tax outcome of these matters is different than the amount recorded, such differences impact income tax expense in the period in which such determination is made. Interest and penalties, if any, related to accrued liabilities for potential tax assessments are included in income tax expense. U.S. GAAP also requires management to evaluate tax positions taken by the Company and recognize a liability if the Company has taken uncertain tax positions that more likely than not would not be sustained upon examination by applicable taxing authorities. Management of the Company has evaluated tax positions taken by the Company and has concluded that as of April 30, 2018, there are no uncertain tax positions taken, or expected to be taken, that would require recognition of a liability that would require disclosure in the financial statements.
| F-9 |
Stock-Based Compensation
The Company accounts for stock option awards in accordance with Financial Accounting Standards Board Accounting Standards Codification (“FASB ASC”) Topic No. 718, Compensation-Stock Compensation. Under FASB ASC Topic No. 718, compensation expense related to stock-based payments is recorded over the requisite service period based on the grant date fair value of the awards. Compensation previously recorded for unvested stock options that are forfeited is reversed upon forfeiture. The Company uses the Black-Scholes option pricing model for determining the estimated fair value for stock-based awards. The Black-Scholes model requires the use of assumptions which determine the fair value of stock-based awards, including the option’s expected term and the price volatility of the underlying stock.
The Company’s accounting policy for equity instruments issued to consultants and vendors in exchange for goods and services follows the provisions of FASB ASC Topic No. 505-50, Equity Based Payments to Non-Employees. Accordingly, the measurement date for the fair value of the equity instruments issued is determined at the earlier of (i) the date at which a commitment for performance by the consultant or vendor is reached or (ii) the date at which the consultant or vendor’s performance is complete. In the case of equity instruments issued to consultants, the fair value of the equity instrument is recognized over the term of the consulting agreement.
Research and Development Expenses
Research and development costs are expensed as incurred. Research and development costs consist of scientific consulting fees and lab supplies, as well as fees paid to other entities that conduct certain research and development activities on behalf of the Company.
The Company has acquired and may continue to acquire the rights to develop and commercialize new product candidates from third parties. The upfront payments to acquire license, product or rights, as well as any future milestone payments, are immediately recognized as research and development expense provided that there is no alternative future use of the rights in other research and development projects.
Loss per Common Share
The Company utilizes FASB ASC Topic No. 260, Earnings per Share. Basic loss per share is computed by dividing loss available to common shareholders by the weighted-average number of common shares outstanding. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common shares had been issued and if the additional common shares were dilutive. Diluted loss per common share reflects the potential dilution that could occur if convertible preferred stock, options and warrants were to be exercised or converted or otherwise resulted in the issuance of Common Stock that then shared in the earnings of the entity.
Since the effects of outstanding options, warrants and convertible preferred stock are anti-dilutive in the period presented, shares of Common Stock underlying these instruments have been excluded from the computation of loss per common share.
| F-10 |
The following sets forth the number of shares of Common Stock underlying outstanding convertible preferred stock, options and warrants:
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Series A convertible preferred stock | 15,000,000 | 27,200,000 | ||||||
| Stock options | 7,500,000 | 7,625,000 | ||||||
| Warrants | 43,000 | 37,500 | ||||||
| 22,543,000 | 34,862,500 | |||||||
Reclassifications
Certain prior period amounts have been reclassified for comparative purposes to conform to the current period financial statement presentation. These reclassifications had no effect on previously reported results of operations.
Recent Accounting Standards
From time to time, new accounting pronouncements are issued by the Financial Accounting Standards Board, (“FASB”), or other standard setting bodies and adopted by the Company as of the specified effective date. Unless otherwise discussed, the impact of recently issued standards that are not yet effective will not have a material impact on the Company’s financial position or results of operations upon adoption.
In August 2016, the FASB issued ASU 2016-15, Statement of Cash Flows (Topic 230) (“ASU 2016-15”), which seeks to reduce the existing diversity in practice in how certain cash receipts and cash payments are presented and classified in the statement of cash flows. For public entities, ASU 2016-15 becomes effective for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years, with early adoption permitted. The Company is currently evaluating the provisions of ASU 2016-15 and assessing the impact, if any, it may have on its financial position, results of operations, cash flows or financial statement disclosures.
In March 2016, the FASB issued ASU 2016-09, Compensation - Stock Compensation (Topic 718) (“ASU 2016-09”), which seeks to simplify accounting for share-based payment transactions including income tax consequences, classification of awards as either equity or liabilities, and the classification on the statement of cash flows. For public entities, ASU 2016-09 becomes effective for fiscal years beginning after December 15, 2016, including interim periods within those fiscal years, with early adoption permitted. The adoption of ASU 2016-09 for the year ended April 30, 2018 did not have a material impact on the Company’s financial position or results of operations.
In January 2016, the FASB issued ASU 2016-01, Recognition and Measurement of Financial Assets and Liabilities (“ASU 2016-01”). ASU 2016-01 requires equity investments (excluding equity method investments and investments that are consolidated) to be measured at fair value with changes in fair value recognized in net income. Equity investments that do not have a readily determinable fair value may be measured at cost, adjusted for impairment and observable price changes. The ASU also simplifies the impairment assessment of equity investments, eliminates the disclosure of the assumptions used to estimate the fair value that is required to be disclosed for financial instruments measured at cost on the balance sheet and requires the exit price to be used when measuring fair value of financial instruments for disclosure purposes. Under ASU 2016-01, changes in fair value (resulting from instrument-specific credit risk) will be presented separately in other comprehensive income for liabilities measured using the fair value option and financial assets and liabilities will be presented separately by measurement category and type either on the balance sheet or in the financial statement disclosures. ASU 2016-01 is effective for fiscal years beginning after December 15, 2017, and interim periods within those fiscal years. The Company has not yet determined the effect that ASU 2016-01 will have on its financial position, results of operations, or financial statement disclosures.
In August 2014, the FASB issued ASU 2014-15, Presentation of Financial Statements-Going Concern (Subtopic 205-40): Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern (“ASU 2014-15”). The amendments in ASU 2014-15 are intended to provide guidance on the responsibility of reporting entity management. Specifically, this ASU 2014-15 provides guidance to management related to evaluating whether there is substantial doubt about the reporting entity’s ability to continue as a going concern and about related financial statement note disclosures. Although the presumption that a reporting entity will continue to operate as a going concern is fundamental to the preparation of financial statements, prior to the issuance of ASU 2014-15, there was no guidance in (U.S. GAAP related to the concept. Due to the lack of guidance in U.S. GAAP, practitioners and their clients often faced challenges in determining whether, when, and how a reporting entity should disclose the relevant information in its financial statements. As a result, the FASB issued this guidance to require management evaluation and potential financial statement disclosures. ASU 2014-15 is effective for financial statements with periods ending after December 15, 2016. The Company adopted ASU 2014-15 during the current fiscal year and determined the effect that ASU 2014-15 did not have a material effect on its financial position, results of operations, or financial statement disclosures.
| F-11 |
In February 2016, the FASB issued No. 2016-02, Leases (“Topic 842” or “ASU 2016-02”), which supersedes the guidance in former ASC 840, Leases. The FASB issued further updates to this guidance in July 2018 through ASU 2018-10, Codification Improvements to Topic 842, Leases and ASU 2018-11, Leases (Topic 842): Targeted Improvements. The new standard requires lessees to apply a dual approach, classifying leases as either finance or operating leases based on the principle of whether or not the lease is effectively a financed purchase by the lessee. This classification will determine whether lease expense is recognized based on an effective interest method or on a straight-line basis over the term of the lease. A lessee is also required to record a right-of-use asset and a lease liability for all leases with a term of greater than 12 months regardless of their classification. Leases with a term of 12 months or less will be accounted for similar to existing guidance for operating leases today. The standard is effective for interim and annual periods beginning after December 15, 2018, with early adoption permitted, and is required to be adopted using a modified retrospective approach. The Company plans to adopt this standard on May 1, 2019. ASU 2016-02 is expected to impact the Company’s consolidated financial statements as the Company has certain operating lease arrangements for which the Company is the lessee. Management is continuing to evaluate the impact the adoption of ASU 2016-02 will have on the Company’s financial position, results of operations and related disclosures. Management expects that the adoption of this standard will result in the recognition of an asset for the right to use a leased facility on the Company’s balance sheet, as well as the recognition of a liability for the lease payments remaining on the lease. While the balance sheet presentation is expected to change, management still does not expect a material change to the condensed consolidated statements of operations and comprehensive loss or cash flows.
In May 2014, the FASB issued ASU No. 2014–09, Revenue from Contracts with Customers (“ASU 2014-09”), which amends the existing accounting standards for revenue recognition. The FASB issued further updates to this guidance through ASU 2016-12 Narrow-Scope Improvements and Practical Expedients, ASU 2016-10 Identifying Performance Obligations and Licensing and ASU 2016-08 Principal Versus Agent Considerations (Reporting Revenue Gross Versus Net). The new standard is based on principles that govern the recognition of revenue at an amount to which an entity expects to be entitled when products are transferred to customers. This standard was adopted on January 1, 2018 using a full retrospective application. There was no impact to the consolidated financial statements upon adoption of ASU 2014-09 as the Company had not recognized any revenue through April 30, 2018.
The Company has considered all other recently issued accounting standards and does not believe the adoption of such standards will have a material impact on its financial statements.
4. NOTE RECEIVABLE, RELATED PARTY, NET
On April 10, 2018, the Company and Avalanche International Corp., a related party (“Avalanche”), entered into a promissory note (the “AVLP Note”) pursuant to which the Company agreed to provide Avalanche a loan of up to $995,500 for a period ending on April 30, 2019, subject to the terms and conditions stated in the AVLP Note. The AVLP Note accrues interest at 10% per annum and includes a 10% original issue discount. At April 30, 2018, the Company has provided loans to Avalanche in the principal amount of $800,000, of which $400,000 was repaid, resulting in net loans to Avalanche of $400,000 and an original issue discount of $40,000.
In accordance with ASC No. 310, Receivables (“ASC 310”), the Company accounts for its AVLP Note at amortized cost, which represents the amount at which the promissory note was acquired, adjusted for accrued interest and accretion of original issue discount. Interest is accreted using the effective interest method. The Company records interest on an accrual basis and recognizes it as earned in accordance with the contractual terms of the promissory note. The original issue discount of $40,000 is being amortized as interest income through the maturity date. During the year ended April 30, 2018, the Company recorded $3,333 of interest income for the discount accretion and recorded contractual interest income from the stated interest rate of $4,008
| F-12 |
5. PREPAID EXPENSES AND OTHER CURRENT ASSETS
Prepaid expenses and other current assets are as follows:
| April 30, 2018 | April 30, 2017 | |||||||
| Prepaid expenses | $ | 1,446,667 | $ | 50,617 | ||||
| Interest receivable | 4,008 | - | ||||||
| Other receivables | 17,010 | 1,700 | ||||||
| Total prepaid expenses and other current assets | $ | 1,467,685 | $ | 52,317 | ||||
Alzamend Consulting Agreement
The Company entered into a five year consulting agreement with Spartan pursuant to which Spartan has agreed to provide consulting services with respect to general corporate matters, including, but not limited to, advice and input with respect to raising capital, potential merger and acquisition transactions, identifying suitable personnel for management, developing corporate structure and finance strategies, assisting the Company with strategic introductions, assisting management with enhancing corporate and shareholder value and introducing the Company to potential investors. In December 2017, since the maximum amount was raised in the Offering, the Company paid to Spartan a consulting fee of $1,400,000 for the services to be rendered over the sixty (60) month term of this consulting agreement.
6. INCOME TAXES
The Company has fully reserved the net deferred income tax assets by taking a full valuation allowance against these assets. As a result of this decision, during the years ended April 30, 2018 and 2017, the Company did not recognize any income tax benefit as a result of its net loss. The table below shows the balances for the deferred income tax assets and liabilities as of the date indicated.
| April 30, 2018 | April 30, 2017 | |||||||
| Deferred income tax asset: | ||||||||
| Net operating loss carryover | $ | 517,972 | $ | 444,110 | ||||
| Other temporary differences | 7,637 | 91,033 | ||||||
| Total deferred tax asset | (25,609 | ) | 535,143 | |||||
| Valuation allowance | 25,609 | (535,143 | ) | |||||
| Deferred income tax asset, net of allowance | $ | - | $ | - | ||||
The income tax provision (benefit) consists of the following:
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Federal and State | ||||||||
| Current | $ | - | $ | - | ||||
| Deferred | 9,543 | (535,143 | ) | |||||
| Valuation allowance | (9,543 | ) | 535,143 | |||||
| Income tax provision (benefit) | $ | - | $ | - | ||||
| F-13 |
On December 22, 2017, the U.S. enacted the Tax Cuts and Jobs Act (“tax reform” or “Tax Act”), which among other things, lowered the U.S. statutory tax rate from 35% to 21% effective January 1, 2018. Consequently, the Company applied a blended U.S. statutory federal income tax rate of 29.7% for fiscal 2018. During the years ended April 30, 2018 and 2017, the Company did not recognize income tax expense. The Company’s effective tax rate was 0% for the years ended April 30, 2018 and 2017. The effective tax rate differed primarily due to the change in the valuation allowance, primarily related to the revaluation of deferred tax assets and liabilities to reflect the new federal tax rate. The reconciliation of income tax attributable to operations computed at the U.S. Federal statutory income tax rate of 29.7% to income tax expense is as follows:
| For the Year Ended April 30, | ||||||||
| 2018 | 2017 | |||||||
| Tax benefit at U.S. Federal statutory tax rate | (29.7 | %) | (34.0 | %) | ||||
| Increase (decrease) in tax rate resulting from: | ||||||||
| Allowance against deferred tax assets | (1.0 | %) | 34.0 | % | ||||
| Nondeductible meals & entertainment expense and other | 0.1 | % | 0.1 | % | ||||
| Taxes in respect of prior years | 0.0 | % | (0.1 | %) | ||||
| Changes in federal tax rate | 30.6 | % | 0.0 | % | ||||
| Effective tax rate | 0.0 | % | 0.0 | % | ||||
At April 30, 2018, the Company had total domestic Federal net operating loss carryovers of approximately $2,467,000 available to offset future taxable income. Federal net operating loss carryovers (“NOLs”) expire beginning in 2026. The Company has not filed its 2016 through 2018 Federal income tax returns. The Company will not be able to utilize these carryovers until the related tax returns are filed. In accordance with Section 382 of the Internal Revenue Code, deductibility of the Company’s NOLs may be subject to an annual limitation in the event of a change of control as defined under the regulations.
In assessing the realization of deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which temporary differences representing net future deductible amounts become deductible. Management considers the scheduled reversal of deferred tax assets, projected future taxable income and tax planning strategies in making this assessment. After consideration of all of the information available and due to the substantial doubt related to the Company’s ability to continue as a going concern and utilize its deferred tax assets, the Company recorded a full valuation allowance of the deferred tax asset. For the year ended April 30, 2018 the valuation allowance has decreased by $9,534.
The 2016 through 2018 tax years remains open to examination by the Internal Revenue Service. The IRS has the authority to examine such tax year until the applicable statute of limitations expire.
7. STOCK-BASED COMPENSATION
On April 30, 2016, the Company’s shareholders approved the Company’s 2016 Stock Incentive Plan (the “Plan”). The Plan provides for the issuance of a maximum of fifty million (12,500,000) shares of the Company’s Common Stock to be offered to the Company’s directors, officers, employees, and consultants. Options granted under the Plan have an exercise price equal to or greater than the fair value of the underlying Common Stock at the date of grant and become exercisable based on a vesting schedule determined at the date of grant. The options expire between 5 and 10 years from the date of grant. Restricted stock awards granted under the Plan are subject to a vesting period determined at the date of grant.
During the years ended April 30, 2018 and 2017, the Company did not grant any equity-based awards from the Plan and did not recognize any stock-based compensation expense from previous grants made pursuant to the Plan. During the year ended April 30, 2017, the Company did issue 337,500 shares of Common Stock to consultants outside of the Plan and recognized $57,447 in stock-based compensation expense the fair value of which was determined from recent sales of the Company’s Common Stock to third parties. Further, pursuant to the terms of the License Agreement, during the years ended April 30, 2018 and 2017, the Company issued 214,967 and 3,386,842 shares of its Common Stock and recognized $218,417 and $56,962, respectively, in license fees.
| F-14 |
All options that the Company grants are granted at the per share fair value on the grant date. Vesting of options differs based on the terms of each option. The Company has valued the options at their date of grant utilizing the Black Scholes option pricing model. As of the issuance of these financial statements, there was not an active public market for the Company’s shares. Accordingly, the fair value of the underlying options was determined based on the historical volatility data of similar companies, considering the industry, products and market capitalization of such other entities. The risk-free interest rate used in the calculations is based on the implied yield available on U.S. Treasury issues with an equivalent term approximating the expected life of the options as calculated using the simplified method. The expected life of the options used was based on the contractual life of the option granted. Stock-based compensation is a non-cash expense because the Company settles these obligations by issuing shares of the Company’s Common Stock from its authorized shares instead of settling such obligations with cash payments.
A summary of option activity under the Company's Plan as of April 30, 2018 and 2017, and changes during the years ended are as follows:
| Outstanding Options | ||||||||||||||||||||
| Weighted | ||||||||||||||||||||
| Weighted | Average | |||||||||||||||||||
| Shares | Average | Remaining | Aggregate | |||||||||||||||||
| Available for | Number of | Exercise | Contractual | Intrinsic | ||||||||||||||||
| Grant | Shares | Price | Life (years) | Value | ||||||||||||||||
| April 30, 2016 | 4,875,000 | 7,625,000 | $ | 0.0004 | 10.00 | $ | - | |||||||||||||
| April 30, 2017 | 4,875,000 | 7,625,000 | $ | 0.0004 | 9.00 | $ | 1,294,804 | |||||||||||||
| Forfeited | 125,000 | (125,000 | ) | $ | 0.0004 | |||||||||||||||
| April 30, 2018 | 5,000,000 | 7,500,000 | $ | 0.0004 | 8.00 | $ | 7,497,000 | |||||||||||||
The aggregate intrinsic value in the table above represents the total pretax intrinsic value (i.e., the difference between the fair value price on the respective date and the exercise price, times the number of shares) that would have been received by the option holders had all option holders exercised their options. There have not been any options exercised during the year ended April 30, 2018.
As of April 30, 2018, there were no unvested stock options and accordingly no unrecognized compensation cost related to unvested stock options.
8. WARRANTS
On October 1, 2017, the Company issued warrants to purchase an aggregate of 5,500 shares of Common Stock at an exercise price equal to $1.20 per share of Common Stock in connection with the issuance of a promissory note in the aggregate principal amount of $44,000 to DPW Holdings, Inc., a related party (See Note 11).
The following table summarizes information about Common Stock warrants outstanding at April 30, 2018:
| Outstanding | Exercisable | ||||||||||||||||||
| Weighted | |||||||||||||||||||
| Average | Weighted | Weighted | |||||||||||||||||
| Remaining | Average | Average | |||||||||||||||||
| Exercise | Number | Contractual | Exercise | Number | Exercise | ||||||||||||||
| Price | Outstanding | Life (years) | Price | Exercisable | Price | ||||||||||||||
| $0.004 | 37,500 | 0.96 | $0.004 | 37,500 | $0.004 | ||||||||||||||
| $1.20 | 5,500 | 2.42 | $1.20 | 5,500 | $1.20 | ||||||||||||||
| $0.004 - $1.20 | 43,000 | 1.14 | $0.156 | 43,000 | $0.156 | ||||||||||||||
| F-15 |
9. RELATED PARTY TRANSACTIONS
As of April 30, 2016, the Company had sold 1,000,000 shares of Series A Convertible Preferred Stock to MCKEA Holdings, LLC (“MCKEA”), a related party (See Note 12).
On May 1, 2016, the Company entered into a Management Services Agreement (“Management Agreement”) with Avalanche International Corp. (“Avalanche”), a related party. The Company’s officers and directors are also officers and directors of Avalanche. Further, MCKEA is the majority member of Philou Ventures, LLC, which is the controlling shareholder of Avalanche. Pursuant to the terms of the Management Services Agreement, Avalanche shall provide management, consulting and financial services to Alzamend. Such services shall include advice and assistance concerning any and all aspects of operations, planning and financing of Alzamend and conducting relations with accountants, attorney, financial advisors and other professionals. The term of the Management Services Agreement, as amended, is for the period May 1, 2016 to December 31, 2017 and may be extended by written agreement. The Company initially paid $40,000 per month for these services and, beginning February 2017, is currently paying $20,000 per month. During the years ended April 30, 2018 and 2017, the Company recognized $240,000 and $420,000, respectively, in management fees. At April 30, 2018 and April 30, 2017, $0 and $180,000, respectively, were included within related party payable on the Company’s balance sheet. The MSA was terminated on December 31, 2018.
On June 28, 2017, MCKEA and Spartan Capital Securities, LLC (“Spartan”) entered into a five-year consulting agreement (the “MCKEA Consulting Agreement”). Pursuant to the MCKEA Consulting Agreement, upon the receipt by the Company of no less than $2,500,000 in gross proceeds from the PPM, MCKEA will transfer to Spartan 5,000,000 shares of Alzamend Common Stock. During the term of the MCKEA Consulting Agreement, Spartan will provide consulting services related to general corporate and other matters related to MCKEA’s investment in the Company such as advice on mergers and acquisition transactions, finance strategies, identification of potential management candidates and other strategic introductions.
The amount due at April 30, 2018 and 2017 to MCKEA and the Company’s officers for reimbursement of expenses paid and incurred by these related parties was $6,636 and $58,373, respectively. The amounts are included within related party payable on the Company’s balance sheet.
The amount were no amounts due at April 30, 2018 for notes payable to related parties. The amount due at April 30, 2017 to related parties from short-term loans, inclusive of unamortized original discount of $6,271, was $253,829. The amounts are included within notes payable, related parties, on the Company’s balance sheet (See Note 11).
On April 10, 2018, the Company and Avalanche International Corp., a related party (“Avalanche”), entered into a promissory note (the “AVLP Note”) pursuant to which the Company agreed to provide Avalanche a loan of up to $995,500 for a period ending on April 30, 2019, subject to the terms and conditions stated in the AVLP Note. The AVLP Note accrues interest at 10% per annum and includes a 10% original issue discount. At April 30, 2018, the Company has provided loans to Avalanche in the principal amount of $800,000, of which $400,000 was repaid, resulting in net loans to Avalanche of $400,000 and an original issue discount of $40,000.
In accordance with ASC No. 310, Receivables (“ASC 310”), the Company accounts for its AVLP Note at amortized cost, which represents the amount at which the promissory note was acquired, adjusted for accrued interest and accretion of original issue discount. Interest is accreted using the effective interest method. The Company records interest on an accrual basis and recognizes it as earned in accordance with the contractual terms of the promissory note. The original issue discount of $40,000 is being amortized as interest income through the maturity date. During the year ended April 30, 2018, the Company recorded $3,333 of interest income for the discount accretion and recorded contractual interest income from the stated interest rate of $4,008 (See Note 4).
10. NOTES PAYABLE
During January 2017, the Company entered into a promissory note and received net proceeds of $65,000. As consideration for this loan, the Company issued a promissory note in the aggregate principal amount of $75,000, which included an OID and fees of $10,000. The OID was amortized as non-cash interest expense over the term of the debt. During the years ended April 30, 2018 and 2017, interest expense of $3,618 and $6,382, respectively was recorded from the debt discount amortization. The promissory note accrued interest at 15% per year and during the years ended April 30, 2018 and 2017, the Company recorded $16,838 and $2,774, respectively, in interest. This loan was repaid during the year ended April 30, 2018. At April 30, 2017, the outstanding balance on the promissory note, inclusive of unamortized OID of $3,618, was $71,382.
| F-16 |
11. NOTES PAYABLE, RELATED PARTY
At April 30, 2017, the outstanding balance on short term borrowings from Avalanche, a related party, was $180,100. During the year ended April 30, 2018, Avalanche provided an additional $123,500 in short term financing. This short-term obligation was non-interest bearing, due upon demand and repaid during the year ended April 30, 2018.
During January 2017, the Company entered a promissory note and received net proceeds of $70,000 from Gary Gottlieb, a related party. As consideration for this loan, the Company issued a promissory note in the aggregate principal amount of $80,000, which included an OID of $10,000. The OID was amortized as non-cash interest expense over the term of the debt. During the years ended April 30, 2018 and 2017, interest expense of $6,271 and $3,729 was recorded from the debt discount amortization. This promissory note accrued interest at 15% per year and during the years ended April 30, 2018 and 2017, the Company recorded $6,272 and $3,614, respectively, in interest. The Company repaid this loan during the year ended April 30, 2018.
On October 1, 2017, the Company entered into a promissory note in the principal amount of $47,520 with DPW Holdings, Inc., a related party (“DPW”). The promissory note included an OID of $3,520 resulting in net proceeds to the Company of $44,000, yielded 8% simple interest on the principal amount, and was due on August 16, 2018. As additional consideration, the Company also issued to DPW a warrant to purchase 5,500 shares of Common Stock at an exercise price of $1.20 per share. The Company recorded debt discount of $2,235 based on the estimated fair value of the 5,500 warrants. Prior to April 30, 2018, the Company repaid the outstanding balance to DPW.
12. EQUITY TRANSACTIONS
Series A Preferred Stock
The Board of Directors has designated 1,360,000 shares of its Preferred Stock as “Series A Convertible Preferred Stock” (the “Series A Preferred Shares”). The Series A Preferred Shares convey no dividend rights except as may be declared by the Board in its sole and absolute discretion, out of funds legally available for that purpose. Holders of Series A Preferred Shares are entitled to 50 non-cumulative votes per share on all matters presented to our stockholders for action. In addition, the affirmative vote of the holders of a majority of the Series A Preferred then outstanding, voting as a separate class, is required for the Company to do any of the following:
| · | amend, alter or repeal any of the preferences or rights of the Series A Preferred Shares; |
| · | authorize any reclassification of the Series A Preferred Shares; |
| · | increase the authorized number of Series A Preferred Shares; or |
| · | create any class or series of shares ranking prior to the Series A Preferred Shares as to dividends or liquidation. |
The Series A Preferred Shares are not entitled to preemptive rights. In the event of any dissolution, liquidation or winding up of the Company, whether voluntary or involuntary, the Holders of Series A Preferred Shares shall be entitled to participate in any distribution out of the assets of the Company on an equal basis per share with the holders of the Common Stock.
Holders of Series A Preferred Shares have the right to convert their shares into shares of Common Stock at any time at a conversion rate equal to twenty (20) shares of Common Stock for every one (1) Series A Preferred Share. The conversion rate is not subject to anti-dilution adjustments.
On April 1, 2016, the Company entered into a subscription agreement with MCKEA Holdings, LLC (“MCKEA”). The Company issued and sold to MCKEA 1,000,000 shares of Series A Preferred Shares that are convertible into 20,000,000 shares of Common Stock that carry the voting power of 50,000,000 shares of Common Stock. Kristine L. Ault, the wife of Milton C. Ault III, Chairman of the Company’s Board of Directors, is the managing member of MCKEA. The issuance resulted in aggregate gross proceeds to the Company of $8,000, of which $5,000 was paid directly to a third party for legal services in April 2016 and $3,000 was received during the year ended April 30, 2017.
| F-17 |
On May 30, 2016, the Company entered into subscription agreements for the sale of 360,000 shares of Series A Preferred Stock to two investors at $0.008/share for an aggregate purchase price of $2,880. The Series A Preferred Stock is convertible into 7,200,000 shares of Common Stock.
Common Stock
On May 27, 2016, the Company’s Board of Directors approved a Certificate of Amendment to the Company’s Certificate of Incorporation increasing its authorized shares of Common Stock from 150,000,000 to 300,000,000.
During April 2016, the Company entered into subscription agreements with multiple investors. The Company issued and sold to these investors 11,600,000 shares of its Common Stock at $.0004 per share. The issuance resulted in aggregate gross proceeds to the Company of $4,640, of which $1,105 was received in April 2016 and the remainder of $3,535 was received in May 2016.
During May 2016, the Company entered into subscription agreements with multiple investors. The Company issued and sold to these investors 11,675,000 shares of its Common Stock at $.0004 per share. The issuance resulted in aggregate gross proceeds to the Company of $4,670, of which $2,970 has been received. The Company has recorded a receivable at October 31, 2017 and April 30, 2017, for the remaining balance due of $1,700 recorded in subscription receivable on the balance sheet.
On June 23, 2016 and July 6, 2016, the Company entered into subscription agreements with EAV, LLC for the purchase of 500 units at $1,000 for each unit purchased. Each unit consisted of 5,875 shares of Common Stock. In aggregate, EAV purchased a total of 1,000 units, representing 5,875,000 shares of Common Stock for an aggregate of $1,000,000, or approximately $0.1704 per share, pursuant to the terms of a Private Placement Memorandum dated June 3, 2016. Payment for the 1,000 units was received between July 7, 2016 and August 2, 2016. In conjunction with the Private Placement Memorandum, the Company incurred $100,000 in placement fees to Palladium Capital Advisors, LLC, and $39,885 in legal and filing fees, resulting in net proceeds to the Company of $860,115.
On April 5, 2016, the Company entered into a retainer agreement for legal representation in connection with a selling shareholder registration on Form 1-A (the “Offering Circular”) for a Regulation A+ offering (the “Reg A+ Offering”). Pursuant to the terms of the retainer agreement the Company agreed to pay a fee of $75,000 of which an initial payment of $25,000 was due upon execution of the retainer agreement, $25,000 is due prior to the filing of the initial Offering Circular and $25,000 is due upon qualification of the Offering Circular by the Securities and Exchange Commission. The total payments of $75,000 are reflected as deferred offering costs, of which $45,000 has been included in accounts payable. During June 2017, the Company raised $74,926 in the Reg A+ Offering, the total amount of the fee will be offset against the proceeds received by reducing additional paid in capital and the balance of $74 will be recorded as legal expense.
An exclusive license agreement with sublicensing terms was made effective on May 1, 2016, as amended on August 17, 2017, (the “Effective Date”) by and between the University, and a direct support organization of the University and the Company (the “License Agreement”). There are certain license fees and milestone payments required to be paid for the licensing of an immunotherapy vaccine peptide that is designed to be used both as a treatment and vaccine against Alzheimer’s (the “Technology”), pursuant to the terms of the License Agreement with the Licensor and the University. Pursuant to the terms of the License Agreement, the Licensor is entitled to receive that number of shares of the Company’s Common Stock equal to five percent (5%) of the total number of issued and outstanding shares outstanding, subject to additional issuances until such time as the Company has received a total of $5 million in cash in exchange for the Company’s equity securities. During the year ended April 30, 2017, the Company issued 3,386,842 shares of its Common Stock and recognized $56,962 in license fees pursuant to the License Agreement. During the year ended April 30, 2018, the Company issued 53,742 shares of its Common Stock and recognized $56,962 in license fees pursuant to the License Agreement. The amount of the license fees was based on the fair value of the Company’s Common Stock on the date of issuance. Fair value was determined from recent sales of the Company’s Common Stock to third parties.
| F-18 |
There are certain initial license fees and milestone payments required to be paid by us to the Licensor pursuant to the terms of the License Agreement. The License Agreement requires the Company to pay royalty payments of four percent (4%) on net sales of products developed from the licensed technology. The Company has already paid an initial license fee of $200,000. As an additional licensing fee, the Licensor is entitled to receive that number of shares of the Company’s Common Stock equal to five percent (5%) of the total number of issued and outstanding shares outstanding on May 1, 2016, subject to additional issuances until such time as the Company has received a total of $5 million in cash in exchange for the Company’s equity securities.
During the year ended April 30, 2017, the Company issued 337,500 shares of Common Stock to service providers for total stock-based compensation of $57,446. All of the shares of Common Stock were valued based on the fair value of the Company’s Common Stock on the date of issuance. Fair value was determined from recent sales of the Company’s Common Stock to third parties. These issuances included 25,000 shares of Common Stock, valued at $4,255, to a related party.
In December 2016, the SEC qualified the Company’s Regulation A Offering Statement pursuant to which the Company sought to raise $50,000,000 from the sale of Common Stock at a price of $8.00 per share. The Company sold 9,366 shares of Common Stock and received gross proceeds of $74,926 in the offering, which closed on June 15, 2017.
On October 19, 2017, the Company entered into subscription agreements for the purchase of 107.7 units at $10,000 for each unit purchased. Each unit consisted of 10,000 shares of Common Stock. In aggregate, the 107.7 units represented 1,077,000 shares of Common Stock for an aggregate purchase price of $1,077,000, or $1.00 per share, pursuant to the terms of a Private Placement Memorandum dated August 17, 2017 (the “PPM”). In conjunction with this PPM, the Company incurred $107,700 in placement fees and $57,900 in legal and filing fees, resulting in net proceeds to the Company of $911,400.
Between December 13, 2017 and December 29, 2017, the Company entered into subscription agreements for the purchase of 311.75 units at $10,000 for each unit purchased. Each unit consisted of 10,000 shares of Common Stock. In aggregate, the 311.75 units represented 3,117,500 shares of Common Stock for an aggregate purchase price of $3,117,500, or $1.00 per share, pursuant to the terms of a Private Placement Memorandum dated August 17, 2017 (the “PPM”). In conjunction with this PPM, the Company incurred $311,750 in placement fees and $35,623 in legal and filing fees, resulting in net proceeds to the Company of $2,770,128.
On January 8, 2018, the Company received notices of conversion from two investors that had purchased 360,000 shares of Series A Preferred Stock. The Series A Preferred Stock was converted into 7,200,000 shares of Common Stock.
Placement Agreement
The Company has agreed with Spartan Capital Securities, LLC (“Spartan”), the placement agent in the Company’s PPM offering (the “Offering”), as follows:
Use of Proceeds
The Company will apply the net proceeds from the Offering to include the retention of a FDA consulting firm, payment of the IND and all associated costs and the launch of a First Stage Clinical Trial with up to 20 human patients along with limited operational expenses.
Corporate Governance
During the period commencing on December 29, 2017 and ending at such time as the Company’s Common Stock is listed on a national securities exchange, Spartan will have the right to designate one member of the Company’s Board of Directors (the “Board”). If Spartan does not elect to designate a member of the Board, then the Company will permit a representative of Spartan to attend all meetings of the Board as an observer.
In addition, commencing within twelve (12) to twenty-four (24) months from December 29, 2017, the Board will be comprised of two inside directors and three independent directors (as such term is defined by Rule 5605 of the NASDAQ Stock Market). This covenant will expire upon the listing of the Company’s Common Stock on a national securities exchange.
| F-19 |
Registration Rights
Subject to applicable law or regulations including but not limited to Rule 415 of the Securities Act the Company, within one hundred and eighty (180) days of the final closing of an initial public offering of the Company’s equity securities, file a registration statement on Form S-1 with the Securities and Exchange Commission, which registration statement will cover the shares of Common Stock issuable to the Placement Agent pursuant to the MCKEA Consulting Agreement discussed above as well as the shares of Common Stock issued in the Offering.
13. SUBSEQUENT EVENTS
In accordance with FASB ASC 855-10, the Company has analyzed its operations subsequent to April 30, 2018 and has determined that it does not have any material subsequent events to disclose in these financial statements except for the following.
On July 2, 2018, the Company obtained two royalty-bearing, exclusive worldwide licenses from the University, to a therapy known as LiProSalTM to mitigate extreme agitation and forestall other deterioration as displayed by patients with up to moderate AD. LiProSalTM is an ionic cocrystal of lithium for the treatment of Alzheimer’s and possibly other neurodegenerative diseases. Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of AD. Unlike traditional medications, which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, it exerts neuroprotective effects, in part, by increasing the brain-derived neurotrophic factor leading to restoration of learning and memory based on the results with some of the animal subjects during the preclinical studies. Another neuroprotective mechanism of lithium is attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Alzamend is continuing to work with two of the inventors of this therapeutic, both at the USF Morsani College of Medicine; Jun Tan, PhD, MD, Professor at the College of Medicine Neurosurgery, Endowed Chair, College of Medicine Psychiatry and Behavioral Neurosciences and the Silver Endowed Chair in Developmental Neurobiology, Department of Psychiatry & Behavioral Medicine and Roland D. Shytle, PhD, Associate Professor, Center of Excellence for Aging & Brain Repair. Dr. Tan and Dr. Shytle with Adam J. Smith, PhD., MSBE, Michael Zaworotko, PhD., and Naga Duggirala, PhD., all formerly with the University of South Florida, have designed, synthesized and characterized this ionic cocrystal of lithium, which Alzamend has named LiProSalTM. LiProSalTM has been shown to exhibit improved pharmacokinetics compared to current FDA-approved lithium-based drugs as well as bioactive in many in vitro models of Alzheimer’s disease. LiProSalTM may prove to be a major improvement over current lithium-based treatments and may also represent a means of treating AD.
There are certain license fees and milestone payments required to be paid for the licensing of the LiProSalTM technology, pursuant to the terms of the Standard Exclusive License Agreements with Sublicensing Terms, both dated June 21, 2018 (the “LiProSalTM License Agreements”) with the Licensor and the University. In addition, a royalty payment of 3% is required pursuant to License #18110 while License #1811 requires a royalty payment of 1.5% on net sales of products developed from the licensed technology. For the two LiProSalTM License Agreements, in the aggregate, the Company is required to pay initial license fees of $50,000 no later than July 31, 2018 and $150,000 no later than October 31, 2018, although the Company is in discussions with the University to defer the payment due date of the initial license fees. As an additional licensing fee, the Licensor is entitled to receive that number of shares of the Company’s Common Stock equal to three percent (3%) of the sum of the total number of issued and outstanding shares. Additionally, the Company is required to pay milestone payments on the due dates to Licensor for the license of the technology, as follows:
| F-20 |
| Payment | Due Date | Event | |||
| $ | 50,000 | November 1, 2019 | Pre-IND meeting | ||
| $ | 65,000 | 6 months from IND filing date | IND application filing | ||
| $ | 190,000 | 12 months from IND filing date | Upon first dosing of patient in a clinical trial | ||
| $ | 500,000 | 12 months from first patient dosing | Upon Completion of first clinical trial | ||
| $ | 1,250,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III Clinical Trial | ||
| $ | 10,000,000 | 8 years from the Effective Date of the Agreement | Upon FDA Approval | ||
On May 29, 2018, the Company implemented a 1-for-4 Reverse Stock Split of its Common Stock. The Reverse Stock Split became effective on December 13, 2018. As a result of the Reverse Stock Split, every four (4) shares of the Company’s pre-Reverse Stock Split Common Stock were combined and reclassified into one share of the Company’s Common Stock. The number of shares of Common Stock subject to outstanding options and warrants were also reduced by a factor of four as of December 13, 2018. All historical share and per-share amounts reflected throughout the consolidated financial statements and other financial information in this filing have been adjusted to reflect the Reverse Stock Split. The par value per share of the Company’s Common Stock was not affected by the Reverse Stock Split.
| F-21 |
Item 8. EXHIBITS
Index to Exhibits
| Exhibit No. | Exhibit Description | |
| 2.1 | Certificate of Incorporation** | |
| 2.2 | Bylaws** | |
| 4.1 | Form of Subscription Agreement** | |
| 6.1 | Standard Exclusive License Agreement with Sublicensing Terms with the University of South Florida Research Foundation, Inc., dated May 1, 2016** | |
| 6.2 | Management Services Agreement, as amended, with Avalanche International Corp., dated May 1, 2016** | |
| 6.3 | Standard Exclusive License Agreement with Sublicensing Terms Number LIC18110 with the University of South Florida Research Foundation, Inc., dated July 2, 2018* | |
| 6.4 | Standard Exclusive License Agreement with Sublicensing Terms Number LIC18111 with the University of South Florida Research Foundation, Inc., dated July 2, 2018* | |
| 8.1 | Escrow Agreement, dated May 24, 2016** |
* Filed herewith
** Previously filed
| 53 |
SIGNATURES
Pursuant to the requirements of Regulation A, the issuer has duly caused this annual report on Form 1-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| Alzamend Neuro, Inc. | |||
| Date: February 21, 2019 | By: | /s/ Stephan Jackman | |
| Stephan Jackman | |||
|
Chief Executive Officer (Principal Executive Officer). |
|||
Pursuant to the requirements of Regulation A, this annual report on Form 1-K has been signed below by the following persons on behalf of the issuer and in the capacities and on the dates indicated.
| Date: February 21, 2019 | By: | /s/ Kenneth S. Cragun | |
| Kenneth S. Cragun | |||
|
Chief Financial Officer (Principal Financial Officer, (Principal Accounting Officer). |