
Filed Pursuant to Rule 424(b)(5)
Registration No. 333-273610
PROSPECTUS SUPPLEMENT
(To Prospectus dated August 10, 2023)
Up to $9,831,947
Alzamend Neuro, Inc.
Shares of Common Stock
We have entered into an At-The-Market Issuance Sales Agreement, or the sales agreement, with Ascendiant Capital Markets, LLC, or ACM, relating to shares of our common stock offered by this prospectus supplement and the accompanying prospectus. In accordance with the terms of the sales agreement, we may offer and sell shares of our common stock, par value $0.0001 per share, having an aggregate offering price of up to $9,831,947 from time to time through ACM, acting as the sales agent, at our discretion.
Our common stock is traded on the Nasdaq Capital Market, or the Exchange, under the symbol “ALZN.” The closing price of our common stock on September 7, 2023 was $0.269 per share.
As of September 8, 2023, the aggregate market value of our outstanding common stock held by non-affiliates, or the public float, was $29,495,841, which was calculated based on 56,635,639 shares of our outstanding common stock held by non-affiliates at a price of $0.5208 per share, the closing price of our common stock on July 19, 2023. Pursuant to General Instruction I.B.6 of Form S-3, in no event will we sell shares pursuant to this prospectus supplement with a value of more than one-third of the aggregate market value of our common stock held by non-affiliates in any 12-month period, or $9,831,947, which period commenced on July 27, 2023, the date we filed our Annual Report on Form 10-K and became subject to the General Instruction I.B.6. of Form S-3, so long as the aggregate market value of our common stock held by non-affiliates is less than $75,000,000.
Sales of our common stock, if any, under this prospectus supplement and accompanying prospectus may be made in sales deemed to be “at the market offerings” as defined in Rule 415 under the Securities Act of 1933, as amended, or the Securities Act. ACM is not required to sell any specific number or dollar amount of securities, but will act as a sales agent using commercially reasonable efforts consistent with its normal trading and sales practices, on terms mutually agreed to by ACM and us. There is no arrangement for funds to be received in any escrow, trust or similar arrangement.
The compensation to ACM for sales of common stock sold pursuant to the sales agreement will be an amount equal to 3% of the gross proceeds of any shares of common stock sold under the sales agreement. In connection with the sale of the common stock on our behalf, ACM may be deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of ACM may be deemed to be underwriting commissions or discounts. We have also agreed to provide indemnification and contribution to ACM with respect to certain liabilities, including liabilities under the Securities Act or the Securities Exchange Act of 1934, as amended, or the Exchange Act.
Investing in our common stock involves a high degree of risk. See “Risk Factors” beginning on page S-23 of this prospectus supplement, on page 11 of the accompanying prospectus and under similar headings in the other documents that are incorporated by reference into this prospectus supplement and the accompanying prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities, or determined if this prospectus supplement or the prospectus to which it relates is truthful or complete. Any representation to the contrary is a criminal offense.
![]()
The date of this Prospectus Supplement is September 8, 2023
TABLE OF CONTENTS
PROSPECTUS SUPPLEMENT
|
Page
| ||
| About this Prospectus Supplement | S-1 | |
| Disclosure Regarding Forward-Looking Statements | S-1 | |
| About the Company | S-3 | |
| The Offering | S-22 | |
| Risk Factors | S-23 | |
| Use of Proceeds | S-47 | |
| Plan of Distribution | S-48 | |
| Description of Our Securities | S-49 | |
| Legal Matters | S-50 | |
| Experts | S-50 | |
| Where You Can Find More Information | S-50 | |
| Incorporation of Documents by Reference | S-50 |
PROSPECTUS
|
Page
| ||
| About this Prospectus | 1 | |
| Disclosure Regarding Forward-Looking Statements | 1 | |
| Prospectus Summary | 2 | |
| Risk Factors | 11 | |
| Use of Proceeds | 11 | |
| Plan of Distribution | 11 | |
| Description of Securities We May Offer | 13 | |
| Description of Capital Stock | 14 | |
| Description of Warrants | 15 | |
| Description of Rights | 17 | |
| Description of Units | 17 | |
| Legal Matters | 18 | |
| Experts | 18 | |
| Where you can find more Information | 18 | |
| Incorporation of Documents by Reference | 19 |
You should rely only on the information contained in this prospectus supplement and the accompanying prospectus. We have not authorized anyone else to provide you with additional or different information. We are offering to sell, and seeking offers to buy, our securities only in jurisdictions where offers and sales are permitted. You should not assume that the information in this prospectus supplement or the accompanying prospectus is accurate as of any date other than the date on the front of those documents or that any document incorporated by reference is accurate as of any date other than its filing date.
No action is being taken in any jurisdiction outside the United States to permit a public offering of our securities or possession or distribution of this prospectus supplement or the accompanying prospectus in that jurisdiction. Persons who come into possession of this prospectus supplement or the accompanying prospectus in jurisdictions outside the United States are required to inform themselves about and to observe any restrictions as to this offering and the distribution of this prospectus supplement and the accompanying prospectus applicable to that jurisdiction.
ABOUT THIS PROSPECTUS SUPPLEMENT
This document is in two parts. The first part is this prospectus supplement, which describes the specific terms of this offering and also adds to and updates information contained in the accompanying prospectus and the documents incorporated by reference into this prospectus supplement and the accompanying prospectus. The second part, the accompanying prospectus, gives more general information about securities we may offer from time to time, some of which does not apply to this offering. Generally, when we refer to this prospectus, we are referring to both parts of this document combined together with all documents incorporated by reference. If the description of the offering varies between this prospectus supplement and the accompanying prospectus, you should rely on the information contained in this prospectus supplement. However, if any statement in one of these documents is inconsistent with a statement in another document having a later date — for example, a document incorporated by reference into this prospectus supplement or the accompanying prospectus — the statement in the document having the later date modifies or supersedes the earlier statement. You should rely only on the information contained in or incorporated by reference into this prospectus supplement or contained in or incorporated by reference into the accompanying prospectus to which we have referred you.
Neither we nor ACM have authorized anyone to provide you with information that is different. If anyone provides you with different or inconsistent information, you should not rely on it. We do not, and ACM does not, take responsibility for, and can provide no assurances as to, the reliability of any information that others provide you. The information contained in, or incorporated by reference into, this prospectus supplement and contained in, or incorporated by reference into, the accompanying prospectus is accurate only as of the respective dates thereof, regardless of the time of delivery of this prospectus supplement and the accompanying prospectus or of any sale of securities. It is important for you to read and consider all information contained in this prospectus supplement and the accompanying prospectus, including the documents incorporated by reference herein and therein, in making your investment decision. You should also read and consider the information in the documents to which we have referred you under the captions “Where You Can Find More Information” and “Incorporation of Documents by Reference” in this prospectus supplement and in the accompanying prospectus.
We are offering to sell, and are seeking offers to buy, the shares only in jurisdictions where such offers and sales are permitted. The distribution of this prospectus supplement and the accompanying prospectus and the offering of the shares in certain jurisdictions or to certain persons within such jurisdictions may be restricted by law. Persons outside the United States who come into possession of this prospectus supplement and the accompanying prospectus must inform themselves about and observe any restrictions relating to the offering of the shares and the distribution of this prospectus supplement and the accompanying prospectus outside the United States. This prospectus supplement and the accompanying prospectus do not constitute, and may not be used in connection with, an offer to sell, or a solicitation of an offer to buy, any securities offered by this prospectus supplement and the accompanying prospectus by any person in any jurisdiction in which it is unlawful for such person to make such an offer or solicitation.
We own or have rights to various trademarks, service marks and trade names that we use in connection with the operation of our business. This prospectus supplement, the accompanying prospectus and the information incorporated herein and thereby by reference may also contain trademarks, service marks and trade names of third parties, which are the property of their respective owners. Our use or display of third parties’ trademarks, service marks, trade names or products in this prospectus supplement or the accompanying prospectus is not intended to, and does not imply a relationship with, or endorsement or sponsorship by us. Solely for convenience, the trademarks, service marks and trade names referred to in this prospectus may appear without the ®, TM or SM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable licensor to these trademarks, service marks and trade names.
Unless otherwise stated or the context requires otherwise, references to “Alzamend,” the “Company,” “we,” “us” or “our” are to Alzamend Neuro, Inc., a Delaware corporation.
DISCLOSURE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference in it contain forward-looking statements regarding future events and our future results that are subject to the safe harbors created under the Securities Act of 1933 and the Securities Exchange Act of 1934. All statements other than statements of historical facts are statements that could be deemed forward-looking statements. These statements are based on our expectations, beliefs, forecasts, intentions and future strategies and are signified by the words “expects,” “anticipates,” “intends,” “believes” or similar language. In addition, any statements that refer to projections of our future financial performance, our anticipated growth, trends in our business and other characterizations of future events or circumstances are forward-looking statements. These forward-looking statements are only predictions and are subject to risks, uncertainties and assumptions that are difficult to predict, including those identified above, under “Risk Factors” and elsewhere in this prospectus. Therefore, actual results may differ materially and adversely from those expressed in any forward-looking statements. All forward-looking statements included in this prospectus are based on information available to us on the date of this prospectus and speak only as of the date hereof.
| S-1 |
We disclaim any current intention to update our “forward-looking statements,” and the estimates and assumptions within them, at any time or for any reason, except as required by U.S. federal securities laws. In particular, the following factors, among others, could cause actual results to differ materially from those described in the “forward-looking statements”:
| • | Our need for substantial additional funding to finance our operations and complete development to seek FDA approval for AL001 and ALZN002 before commercialization; | |
| • | Our ability to effectively execute our business strategy; | |
| • | Our ability to manage our expansion, growth and operating expenses; | |
| • | Our ability to evaluate and measure our business, prospects and performance metrics; | |
| • | Our ability to compete and succeed in a highly competitive and evolving industry; | |
| • | Our ability to respond and adapt to changes in technology and customer behavior; | |
| • | Our ability to protect our intellectual property and to develop, maintain and enhance a strong brand; | |
| • | Our significant losses since inception and anticipation that we will continue to incur significant losses for the foreseeable future; | |
| • | Our reliance on licenses from a third party regarding our rights and development of AL001 and AL002; | |
| • | Our development of AL001 and AL002 never leading to a marketable product; | |
| • | Our product candidates not qualifying for expedited development, or if they do, not actually leading to a faster development or regulatory review or approval process; | |
| • | Our approach to targeting beta-amyloid plaque via AL002 being based on a novel therapeutic approach; and | |
| • | The risk factors included in our most recent filings with the SEC, including, but not limited to, our Forms 10-K and 10-Q, which are incorporated by reference herein. |
| S-2 |
ABOUT THE COMPANY
This summary highlights selected information contained in other parts of this prospectus. Because it is a summary, it does not contain all of the information that you should consider in making your investment decision. Before investing in our securities, you should read the entire prospectus carefully, including the information set forth under the heading “Risk Factors.”
Company Overview
Alzamend Neuro, Inc., a Delaware corporation, was incorporated in February 2016 (sometimes referred to as “Alzamend,” the “Company,” “we” or “us”). We are a clinical-stage biopharmaceutical company focused on developing novel products for the treatment of Alzheimer’s disease (“Alzheimer’s”), bipolar disorder (“BD”), major depressive disorder (“MDD”) and post-traumatic stress disorder (“PTSD”). With our two product candidates, we aim to bring treatments or potential cures to market as quickly as possible. Far too many individuals, patients and caregivers suffer from the burden created by these devastating, and often fatal, diseases. Our primary target, Alzheimer’s, was among the most-feared diseases (second only to cancer) among Americans, according to a 2011 survey by the Harvard School of Public Health. Alzheimer’s is also the seventh leading cause of death in the United States (“U.S.”) according to a 2021 report from the Alzheimer’s Association, a nonprofit that funds research. Existing Alzheimer’s treatments only temporarily relieve symptoms and while one treatment has been shown to slow the progression of the disease, no treatments have been shown to halt the progression of the disease, which currently affects roughly 6.7 million Americans and that number is expected to grow to 13 million individuals by 2050. Alzheimer’s also impacts more than 11 million Americans who provide an estimated 16 billion hours of unpaid care per year, valued at $272 billion, according to data provided by the Alzheimer’s Association. In 2022, the estimated healthcare costs for treating individuals with Alzheimer’s in the U.S. will be $321 billion, including $206 billion in Medicare and Medicaid payments. These costs could rise to as high as $1 trillion per year by 2050 if no permanent treatment or cure for Alzheimer’s is found, the Alzheimer’s Association reported.
Our pipeline consists of two novel therapeutic drug candidates:
| · | AL001 - A patented ionic cocrystal technology delivering a therapeutic combination of lithium, salicylate and proline through three royalty-bearing exclusive worldwide licenses from the University of South Florida Research Foundation, Inc., as licensor (the “Licensor”); and |
| · | ALZN002 - A patented method using a mutant peptide sensitized cell as a cell-based therapeutic vaccine that seeks to restore the ability of a patient’s immunological system to combat Alzheimer’s through a royalty-bearing exclusive worldwide license from the Licensor. |
Our most advanced product candidate (lead product) is licensed and in clinical development in humans is AL001, an ionic cocrystal of lithium for the treatment of Alzheimer’s, BD, MDD and PTSD. Based on our preclinical data involving mice models, AL001 treatment prevented cognitive deficits, depression and irritability and is superior in improving associative learning and memory and irritability compared with lithium carbonate treatments, supporting the potential of AL001 for the treatment of Alzheimer’s, BD, MDD and PTSD in humans. Lithium has been marketed for more than 35 years and human toxicology regarding lithium use has been well characterized, potentially mitigating the regulatory burden for safety data.
The results of randomized, placebo-controlled, clinical trials of lithium in the treatment of patients with Alzheimer’s dementia and subjects with mild cognitive impairment have been widely published. Clinical studies have indicated that lithium administered at doses lower than those used for affective disorders can favorably impact Alzheimer’s outcomes. A study by O.V. Forlenza, et al., entitled “Disease-Modifying Properties of Long-Term Lithium Treatment for Amnestic Mild Cognitive Impairment: Randomized Controlled Trial,” appearing in the British Journal of Psychiatry (2011) reported that lithium was superior to a placebo, evidencing a slower decline of cognitive function as measured by the Alzheimer’s Disease Assessment Scale cognitive subscale. Given the absence of adequate, widely adapted treatments that can slow, halt or even reverse the decline of this highly prevalent disease, the potential efficacy of lithium in the long-term management of Alzheimer’s may positively impact public health. There is an unmet medical need for safe and effective Alzheimer’s treatments, particularly for treatments with neuroprotective properties.
There is increasing evidence to suggest that depressive illness, particularly in the elderly, is associated with neuronal cell loss. These findings suggest that lithium may exert some of its long-term beneficial effects in the treatment of affective disorders via underappreciated neuroprotective effects. Molecular biology and animal studies have also suggested that lithium may offer protection against Alzheimer’s. Given the absence of other adequate treatments, the potential efficacy of lithium in the long-term treatment of neurodegenerative disorders may be warranted.
Our Business Strategy
We intend to develop and commercialize therapeutics that are better than existing treatments and have the potential to significantly improve the lives of individuals afflicted by Alzheimer’s, BD, MDD and PTSD. To achieve these goals, we are pursuing the following key business strategies:
| S-3 |
| • | Advance clinical development of AL001 for Alzheimer’s, BD, MDD and PTSD treatment. We completed our Phase I clinical trial in March 2022 and initiated a Phase IIA MAD clinical trial in May 2022. We completed the clinical portion of the Phase IIA Multiple Ascending Dose (“MAD”) clinical trial in March 2023 and reported topline data in June 2023. We intend to initiate two Phase II clinical trials to investigate the safety and efficacy of AL001 for patients with mild to moderate Alzheimer’s. Additionally, we intend to investigate the potential of AL001 for patients suffering from BD, MDD and PTSD by submitting investigational new drug (“IND”) applications to the U.S. Food and Drug Administration (“FDA”) for these indications by the end of 2023. If we achieve successful Phase III clinical trials in humans, we intend to seek approval to commercialize AL001 via a New Drug Application (“NDA”); |
| • | Advance clinical development of ALZN002 for Alzheimer’s treatment. We submitted an IND application to the FDA in September 2022, and received a “study may proceed” letter in October 2022. In April 2023, we initiated a Phase I/IIA clinical trial for ALZN002 to treat mild to moderate dementia of the Alzheimer’s type. If we achieve successful Phase III clinical trials in humans, we intend to seek approval to commercialize ALZN002 via an NDA; |
| • | Expand our pipeline of pharmaceuticals to include additional indications for AL001 and delivery methods. Another element of our business strategy is to expand our pipeline of pharmaceuticals based on our technology and advance these product candidates through clinical development for the treatment of a variety of indications. In addition to treating Alzheimer’s, AL001 has the potential to treat a wide range of neurodegenerative diseases and psychiatric disorders. We plan to pursue the treatment of BD, MDD, and PTSD with AL001, and in May 2022, we submitted a pre-Investigational New Drug (“pre-IND”) meeting request to the FDA for these indications and received a written response from the FDA in July 2022. Based on the written response from the FDA and the receipt of topline data from the Phase IIA MAD clinical trial, we plan to submit separate INDs for BD, MDD, and PTSD by the end of 2023, which, after receipt from the FDA of a “study may proceed” letter for such indication, would allow us to initiate a Phase II study. We also plan to explore different formulations (liquid, immediate release and sprinkle capsules) to deliver AL001; |
| • | Focus on translational and functional endpoints to efficiently develop product candidates. We believe AL001 is positioned for a Section 505(b)(2) regulatory pathway for new drug approvals. We also believe AL001 and ALZN002 are positioned for breakthrough therapy designations because of their positive effects on a pharmacodynamic biomarker (beta-amyloids) and potential for a clinically meaningful effect on Alzheimer’s, making them eligible to receive assistance from the FDA throughout the development process that may shorten the development timelines. However, we have neither received breakthrough therapy designation nor have we qualified for expedited development, and no assurance can be given that we will. Even if we qualify for breakthrough therapy designation or expedited development, it may not actually lead to faster development or expedited regulatory review and approval or necessarily increase the likelihood that we will receive FDA approval; and |
| • | Optimize the value of AL001 and ALZN002 in major markets. We intend to commercialize AL001 and ALZN002 by seeking FDA marketing approval for both product candidates and partnering with biopharmaceutical companies seeking to strategically fortify pipelines and, in turn, receiving funding for the costly later-stage clinical development. We do not anticipate selling products directly into the marketplace, though we may do so depending on market conditions. Our focus is expected to concentrate on entering into strategical transactions with established distributors and producers, which will provide distribution and marketing capabilities for the sale of our products into the marketplace. |
Our Development Pipeline
The following chart provides an overview of the current development stages of our therapeutic product candidates.
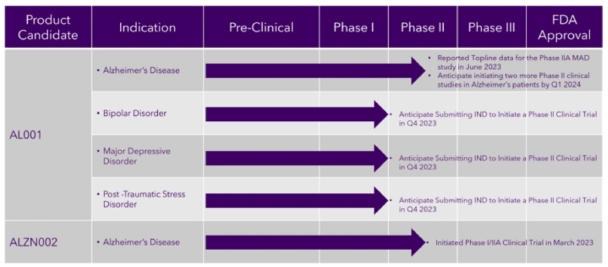
| S-4 |
Our product candidates will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it or any successors are likely to provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval for and commercialize our product candidates, our long-term business plans will not be met, and we will be unable to generate the revenue we have forecast for the foreseeable future, if any. We do not anticipate that we will generate our maximum revenue for several years, or that we will achieve profitability for any of our therapeutic drug candidates until at least a few years after generating material revenue, if at all. If we are unable to generate revenue or raise substantial additional capital, we will not be able to pursue any expansion of our business or acquire additional intellectual property, we will not become profitable with our therapeutic drug candidates, and we will be unable to continue our operations at the currently planned pace, if at all.
AL001 Drug Candidate
Our lead product candidate that we have licensed and have begun clinical development in humans is an ionic cocrystal of lithium for the treatment of Alzheimer’s, BD, MDD and PTSD. Lithium salts have a long history of human consumption beginning in the 1800s. In psychiatry, they have been used to treat mania and as a prophylactic for depression since the mid-20th century. Today, lithium salts are used as a mood stabilizer for the treatment of BD. Although the FDA has approved no medications as safe and effective treatments for suicidality, lithium has proven to be the only drug that consistently reduces suicidality in patients with neuropsychiatric disorders. Despite these effective medicinal uses, current FDA-approved lithium pharmaceutics (lithium carbonate and lithium citrate) are limited by a narrow therapeutic window that requires regular blood monitoring of plasma lithium levels and blood chemistry by a clinician to mitigate adverse events. Because conventional lithium salts (carbonate and citrate) are eliminated relatively quickly, multiple administrations throughout the day are required to safely reach therapeutic plasma concentrations. Existing lithium drugs, such as lithium chloride and lithium carbonate, suffer from chronic toxicity, poor physicochemical properties and poor brain bioavailability. Because lithium is so effective at reducing manic episodes in patients with BD, it is still used clinically despite its narrow therapeutic index. This has led researchers to begin to look for alternatives to lithium with similar bioactivities.
Scientists from the University of South Florida have developed a new lithium cocrystal composition and method of preparation that, under certain clinical and/or testing conditions, have been shown to allow for lower dosages to achieve therapeutic brain levels of lithium for psychiatric disorders, which could lead to a broadening of lithium’s therapeutic index. Our studies and/or testing have indicated that the compound offers improved physiochemical properties compared to existing forms of lithium, giving it the potential to be developed as an anti-suicidal drug and for use against mood disorders.
Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of Alzheimer’s. Unlike traditional medications which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, recent studies have indicated that it exerts neuroprotective effects, in part, by increasing a brain-derived neurotrophic factor leading to restoration of learning and memory. Another neuroprotective mechanism of lithium indicated by recent studies is the attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Results from recent clinical studies suggest that lithium treatment may reduce dementia development while preserving cognitive function and reducing biomarkers associated with Alzheimer’s.
The novel ionic cocrystal of lithium (AL001), which was designed, synthesized and characterized by a team of inventors from the University of South Florida has been shown to exhibit improved nonclinical pharmacokinetics compared to currently FDA-approved lithium products and is also bioactive in many in vitro models of Alzheimer’s. AL001 may constitute a means of treating Alzheimer’s, BD, MDD and PTSD.
We believe that our ability to re-engineer lithium solid dosage forms in order to optimize performance and has the potential to address a wide range of clinical applications ranging from neurodegenerative disorders, not merely Alzheimer’s, but also amyotrophic lateral sclerosis (known as ALS and Lou Gehrig’s disease), Huntington’s disease, multiple sclerosis, Parkinson’s disease and traumatic brain injury, to more psychiatric conditions such as BD, MDD, mania, PTSD and suicidality. This novel approach is intended to achieve the desired therapeutic outcome of enhanced penetration through the blood-brain barrier and sustained brain lithium concentrations while systemic exposures (and toxicities) are mitigated for other organ systems. The optimal modified-release lithium dosing approach for AL001 should avoid acutely toxic peak concentrations in blood, as well as in the brain, and should maintain such blood concentrations for a predictable, clinically relevant time, with overall low systemic exposures that mitigate the potential for adverse events. We anticipate that the lithium delivery system will be adaptable to a dosing regimen that maintains therapeutic brain lithium concentrations consistently for the longest possible time while allowing only modest exposures and providing adequate recovery periods between doses for other organ systems.
| S-5 |
Clinical Trials
Phase I Study
On September 13, 2021, we initiated a randomized, balanced, Phase I, single-dose, open-label, two-treatment, two-period, two- sequence, crossover, relative bioavailability clinical trial to investigate lithium pharmacokinetics and safety of AL001 formulation compared to a marketed immediate release lithium carbonate formulation in healthy subjects. The primary objective of this clinical trial was to assess the relative bioavailability of the AL001 lithium formulation relative to a marketed lithium carbonate formulation in healthy subjects for the purpose of determining potential clinically safe and effective AL001 dosing in future studies. Additionally, we wanted to characterize safety and tolerability of the tested formulations under the conditions of this clinical trial. This was a first-in-human clinical trial of the AL001 formulation; this trial was designed to assess the relative bioavailability of the AL001 lithium formulation compared to a marketed lithium carbonate formulation in at least 24 completed healthy subjects (30 subjects were to be enrolled) for the purpose of determining potential clinically safe and effective AL001 dosing in future clinical trials. The AL001 lithium content was nearly half of the reference lithium carbonate capsule dosage as it was expected that treatment of frail Alzheimer’s patients will require half the lithium dose used for treatment of BD. Lithium carbonate 300 mg (Reference product) was given as a single dose in this clinical trial; this is often used as a starting dose for treatment of BD when given three times daily. The shape of the AL001 lithium plasma concentration versus time curve was unknown prior to this study. Also unknown were the AL001 rate and extent of lithium absorption. The Phase I study was completed in March 2022 with the following results:
| · | AL001 was shown to be safe and well-tolerated in healthy adult subjects; |
| · | No serious adverse events and no deaths were reported during the trial; |
| · | The safety profiles of both AL001 and the marketed lithium carbonate capsule were benign; |
| · | No clinically significant abnormal findings in electrocardiograms were noted during the trial; |
| · | AL001 salicylate plasma concentrations were observed to be well tolerated and consistently within safe limits; and |
| · | Dose-adjusted relative bioavailability analyses of the rate and extent of lithium absorption in plasma indicated that AL001, at a lithium carbonate equivalent dose of 150 mg, is bioequivalent to a marketed 300 mg lithium carbonate capsule and the shapes of the lithium plasma concentration versus time curves are similar. |
Phase IIA Study
On May 5, 2022, we initiated a multiple-dose, steady-state, double-blind, ascending dose safety, tolerability, pharmacokinetic clinical trial (www.clinicaltrials.gov, identifier: NCT05363293) of AL001 in patients with mild to moderate Alzheimer’s and healthy subjects with the following objectives:
| · | Primary: To evaluate the safety and tolerability of AL001 under multiple-dose, steady-state conditions in Alzheimer’s patients and healthy subjects; |
| · | Secondary: To characterize the maximum tolerated dose (MTD) of AL001 in patients with mild to moderate Alzheimer’s and healthy subjects; and |
| · | Exploratory: Determination of qualitative and quantitative evaluations of AD patient and healthy subjects desirable characteristics for future Phase II and III clinical studies in order to: |
| o | Facilitate recruitment into subsequent AL001 clinical trials; and |
| o | Facilitate trial-adherence to completion of study requirements including treatment adherence. |
We completed the Phase IIA clinical trial in March 2023 and announced positive topline data in June 2023. We announced that we successfully identified a maximum tolerated dose (“MTD”) for development of AL001 from a multiple-ascending dose study as assessed by an independent safety review committee. This dose, providing lithium at a lithium carbonate equivalent dose of 240 mg 3-times daily (“TID”), is designed to be unlikely to require lithium therapeutic drug monitoring (“TDM”). Also, this MTD is risk mitigated for the purpose of treating fragile populations, such as Alzheimer’s patients.
Lithium is a commonly prescribed drug for manic episodes in BP type 1 as well as maintenance therapy of BP in patients with a history of manic episodes. Lithium is also prescribed off-label for MDD, BP and treatment of PTSD, among other disorders. Lithium was the first mood stabilizer approved by the FDA and is still a first-line treatment option (considered the “gold standard”) but is underutilized perhaps because of the need for TDM. Lithium was the first drug that required TDM by regulatory authorities in product labelling because the effective and safe range of therapeutic drug blood concentrations is narrow and well defined for treatment of BP when using lithium salts. Excursions above this range can be toxic, and below can impair effectiveness.
| S-6 |
Planned Future Studies
Based on the results from our Phase IIA MAD study, we plan to initiate two safety and efficacy clinical trials in subjects with mild to moderate dementia of the Alzheimer’s type. Additionally, we intend to investigate the potential of AL001 for patients suffering from BD, MDD and PTSD by submitting IND applications to the FDA for these indications by the end of 2023. After FDA permission to proceed on the INDs, we intend to initiate clinical trials at this MTD to determine relative increased lithium levels in the brain compared to a marketed lithium salt for BD, MDD and PTSD, based on published mouse studies that predict that lithium can be given at lower doses for equivalent therapeutic benefit when treating with AL001. For example, the goal is to replace a 300 mg TID lithium carbonate dose for treatment of BD with a 240 mg TID AL001 lithium equivalent, which represents a daily decrease of 20% of lithium given to a patient.
ALZN002 Drug Candidate
The other product candidate that we have licensed to clinically develop in humans is ALZN002, a patented method using a mutant peptide sensitized cell as a cell-based therapeutic vaccine which seeks to restore the ability of the patient’s immunological system to combat Alzheimer’s. The proposed mechanism of action is through the pulsed-Dendritic Cell (“DC”) activation of T-cells that stimulates the immune system, resulting in the clearance of brain amyloid. Preclinical studies conducted from April 2005 to July 2010 demonstrated that the infusion of transgenic (or genetically modified) mice with ALZN002-pulsed DCs is associated with lower amyloid burden and improved neuro-behavioral performance. This is likely to be mediated by an anti-inflammatory effect in addition to the immunogenicity of this therapy.
ALZN002 is based on the theory that Alzheimer’s symptoms may be caused in large part by plaque deposits that can cluster in the brain composed of protein fragments called beta-amyloids that build up between nerve cells. One hypothesis is that a special type of immune cell, natural beta-amyloid antibodies, may play a role in preventing plaque build-up in people without Alzheimer’s. As people age, their immune systems may degrade, and some people may be unable to produce natural beta-amyloid antibodies, the absence of which leads to the plaque build-up causing Alzheimer’s.
ALZN002 is intended to elicit an immune response to produce anti-amyloid antibodies, which can then neutralize circulated beta-amyloids and prevent additional plaque build-up. The mutant antigen within ALZN002 was selected specifically for its high Human Leukocyte Antigens (“HLA”) binding affinity, thereby avoiding the need for an adjuvant, which may cause an adverse (Th1) immune response.
ALZN002 is an autologous modified DC treatment. More precisely, it is a patient-specific therapy where the patient undergoes leukapheresis, a nonsurgical treatment used to reduce the quantity of white blood cells in the bloodstream, to isolate peripheral blood monocytes that are subsequently matured into DCs using cytokine therapy (IL4+ GM-CSF) cocktail. The DCs are incubated with a modified amyloid beta (Aβ) peptide to sensitize them, and then administered to the same patient.
Significant evidence has accumulated recently suggesting that immunotherapy is a highly promising modality of treatment in Alzheimer’s. Most current immune-based active investigations are focused on passive immunization by pre-prepared Aβ antibody administration. Active immunization may offer additional or more lasting effects on the clearance of amyloid and a safer approach due to its reliance on autologous immune mechanisms. Further, preliminary evidence suggests a recurrence of the amyloid accumulation after clearance with the immunoglobulins. A prior attempt at engaging the immune system to treat Alzheimer’s was conducted using the immunization with pre-aggregated synthetic Aβ (AN-1792) combined with the immunogenic adjuvant QS-21. The Phase IIA study with AN-1792 was terminated by the FDA due to severe meningoencephalitis in approximately 6% of vaccinated subjects. We believe that this may have been caused by using a QS-21 adjuvant in the vaccine formulation.
Clinical Trials
Pre-Clinical
On July 23, 2021, we announced that Alzamend received positive toxicology results for ALZN002 in a good laboratory practices (“GLP”) toxicology study using a transgenic mouse model of Alzheimer’s. The study was conducted by Charles River Laboratories. ALZN002 is a patented method using a mutant-peptide sensitized cell as a cell-based therapeutic vaccine that seeks to restore the ability of a patient’s immunological system to combat Alzheimer’s.
A five-dose GLP study with ALZN002-sensitized cells was completed using a transgenic (or genetically modified) mouse model of Alzheimer’s to investigate the tolerability of ALZN002. Single injections were administered on days 1, 30, 50, 70, and 90. The mice were evaluated for potential toxicity and reversibility of any findings at 75 and 90 days after dosing.
Histopathology results demonstrate that there was no indication of T-cell infiltration or meningoencephalitis suggesting that ALZN002 therapy is safe and tolerable as there were no adverse findings over a 90-day period and 90 days after the last dose. There were no treatment-related mortalities or reports of adverse effects on clinical observations, body weight parameters, organ weight parameters, clinical pathology parameters, gross pathology observations, or histopathologic observations during the main study or the recovery phase.
| S-7 |
Modified cell therapies, especially DCs, may provide a safer and more patient-specific active immunization. Ex-vivo modification of DCs as a modality of treatment has been previously used in oncological therapeutics. It has been shown to be relatively safe and capable of engaging the immune system to attack the target tissues with success. Its use in Alzheimer’s therapeutics is relatively recent.
Phase I/II Study
We submitted a pre-IND meeting request for ALZN002 and supporting briefing documents to the Center for Biological Evaluation and Research of the FDA on July 30, 2021. We received a written response relating to the pre-IND from the FDA providing a path for Alzamend’s planned clinical development of ALZN002 on September 30, 2021. The FDA agreed to allow Alzamend to submit an IND to conduct a combined Phase I/II study.
On September 28, 2022, we submitted an IND application to the FDA for ALZN002 and received a “study may proceed” letter on October 31, 2022. The product candidate is an immunotherapy vaccine designed to treat mild to moderate dementia of the Alzheimer’s type. ALZN002 is a proprietary “active” immunotherapy product, which means it is produced by each patient’s immune system. It consists of autologous DCs that are activated white blood cells taken from each individual patient so that they can be engineered outside of the body to attack Alzheimer’s-related amyloid-beta proteins. These DCs are pulsed with a novel amyloid-beta peptide (E22W) designed to bolster the ability of the patient’s immune system to combat Alzheimer’s; the goal being to foster tolerance to treatment for safety purposes while stimulating the immune system to reduce the brain’s beta-amyloid protein burden, resulting in reduced Alzheimer’s signs and symptoms. Compared to passive immunization treatment approaches that use foreign blood products (such as monoclonal antibodies), active immunization with ALZN002 is anticipated to offer a more robust and long-lasting effect on the clearance of amyloid. This could provide a safer approach due to its reliance on autologous immune components, using each individual patient’s own white blood cells rather than foreign cells and/or blood products.
On April 3, 2023, we announced the initiation of a phase I/IIA clinical trial for ALZN002 to treat mild to moderate dementia of the Alzheimer’s type. The purpose of this trial is to assess the safety, tolerability, and efficacy of multiple ascending doses of ALZN002 compared with that of a placebo in 20-30 subjects with mild to moderate morbidity. The primary goal of this clinical trial is to determine an appropriate dose of ALZN002 for treatment of patients with Alzheimer’s in a larger Phase IIB efficacy and safety clinical trial, which Alzamend expects to initiate within three months of receiving data from the initial trial.
The continuation of our current development plans with respect to completing our IND applications and conducting the series of human clinical trials for each of our therapeutics requires us to raise additional capital to fund our operations.
Intellectual Property and Licensing Agreements
On July 2, 2018, we entered into two Standard Exclusive License Agreements with Sublicensing Terms for AL001 with the Licensor and its affiliate, the University of South Florida (the “AL001 Licenses”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide licenses limited to the field of Alzheimer’s, under U.S. Patent Nos. (i) 9,840,521, entitled “Organic Anion Lithium Ionic Cocrystal Compounds and Compositions”, filed September 24, 2015 and granted December 12, 2017, and (ii) 9,603,869, entitled “Lithium Co-Crystals for Treatment of Neuropsychiatric Disorders”, filed May 21, 2016 and granted March 28, 2017. On February 1, 2019, we entered into the First Amendment to the AL001 Licenses, on March 30, 2021, we entered into the Second Amendment to the AL001 Licenses and on June 8, 2023, we entered into the Third Amendment to the AL001 Licenses (collectively, the “AL001 License Agreements”).
The AL001 License Agreements require that we pay combined royalty payments of 4.5% on net sales of products developed from the licensed technology for AL001. We have already paid an initial license fee of $200,000 for AL001. As an additional licensing fee for the license of the AL001 technologies, the Licensor received 2,227,923 shares of our common stock. Minimum royalties for AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the AL001 License Agreements.
On May 1, 2016, we entered into a Standard Exclusive License Agreement with Sublicensing Terms for ALZN002 with the Licensor (the “ALZN002 License”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide license limited to the field of Alzheimer’s Immunotherapy and Diagnostics, under U.S. Patent No. 8,188,046, entitled “Amyloid Beta Peptides and Methods of Use”, filed April 7, 2009 and granted May 29, 2012. On August 18, 2017, we entered into the First Amendment to the ALZN002 License, on May 7, 2018, we entered into the Second Amendment to the ALZN002 License, on January 31, 2019, we entered into the Third Amendment to the ALZN002 License, on January 24, 2020, we entered into the Fourth Amendment to the ALZN002 License, on March 30, 2021, we entered into the Fifth Amendment to the ALZN002 License and on April 17, 2023, we entered into the Sixth Amendment to the ALZN002 License (collectively, the “ALZN002 License Agreement”).
| S-8 |
The ALZN002 License Agreement requires us to pay royalty payments of 4% on net sales of products developed from the licensed technology for ALZN002. We have already paid an initial license fee of $200,000 for ALZN002. As an additional licensing fee for the license of ALZN002, the Licensor received 3,601,809 shares of our common stock. Minimum royalties for ALZN002 are $20,000 on the first anniversary of the first commercial sale, $40,000 on the second anniversary first commercial sale and $50,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the ALZN002 License Agreement.
On November 19, 2019, we entered into two Standard Exclusive License Agreements with Sublicensing Terms for two additional indications of AL001 with the Licensor (the “November AL001 License”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide licenses limited to the fields of (i) neurodegenerative diseases excluding Alzheimer’s and (ii) psychiatric diseases and disorders. On March 30, 2021, we entered into the First Amendments to the November AL001 License and on April 17, 2023, we entered into the Second Amendments to the November AL001 License (collectively, the “November AL001 License Agreements”).
The November AL001 License Agreements require us to pay royalty payments of 3% on net sales of products developed from the licensed technology for AL001 in those fields. We paid an initial license fee of $20,000 for the additional indications. Minimum royalties for November AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the November AL001 License Agreements.
These license agreements have an indefinite term that continue until the later of the date no licensed patent under the applicable agreement remains a pending application or enforceable patent, the end date of any period of market exclusivity granted by a governmental regulatory body, or the date on which the licensee’s obligations to pay royalties expire under the applicable license agreement. Under our various license agreements, if we fail to meet a milestone by its specified date, Licensor may terminate the license agreement. The Licensor was also granted a preemptive right to acquire such shares or other equity securities that may be issued from time to time by us while the Licensor remains the owner of any equity securities of our company.
Additionally, we are required to pay milestone payments on the due dates to the Licensor for the license of the AL001 technologies and for the ALZN002 technology, as follows:
Original AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Completed September 2019 | Pre-IND meeting | |
| $ | 65,000 | * | Completed June 2021 | ND application filing | |
| $ | 190,000 | * | Completed December 2021 | Upon first dosing of patient in a clinical trial | |
| $ | 500,000 | * | Completed March 2022 | Upon Completion of first clinical trial | |
| $ | 1,250,000 | 24 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 8 years from the effective date of the agreement | Upon FDA NDA approval | ||
| * | Milestone met and completed |
| S-9 |
ALZN002 License:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Upon IND application filing | Upon IND application filing | |
| $ | 50,000 | September 2023 | Upon first dosing of patient in first Phase I clinical trial | ||
| $ | 500,000 | 24 months from completion of first Phase I clinical trial | Upon completion of first Phase II clinical trial | ||
| $ | 1,000,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA Biologics License Application (“BLA”) approval | ||
| * | Milestone met and completed |
Additional AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 2,000,000 | 36 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 16,000,000 | August 1, 2029 | First commercial sale | ||
Industry Overview
Alzheimer’s
Currently, Alzheimer’s is the seventh leading cause of death in the U.S. and, when extrapolated globally, the market for preventions, treatments and cures of this crippling disease is massive. Since 1990, life expectancy has increased by six years and the worldwide average continues to increase. With the increase in the mean age of the population in developed countries, the prevalence of deteriorating neurological diseases has also increased. According to the Alzheimer’s Association, in the U.S. alone, one of nine persons over the age of 65 have Alzheimer’s, with roughly 6.7 million Americans currently living with it. It is estimated that this number will grow to 13 million by 2050 barring the development of medical breakthroughs to prevent, slow or cure the disease. Many Alzheimer’s related associations believe the actual number of adults with Alzheimer’s may be much higher since current statistics do not account for deaths from complications or from related diseases like pneumonia or heart attack. These death certificates only list the most immediate cause. The fastest growing age group in the U.S. is the “over 85” group within which one in three individuals have Alzheimer’s.
It is estimated that the cost of caring for people with Alzheimer’s and other dementias will increase from an estimated $345 billion in 2023 to a projected $1 trillion per year by 2050 with Medicare and Medicaid covering approximately 70% of such costs. Over 11 million Americans provide unpaid care for people with Alzheimer’s or other dementias. The Alzheimer’s Association estimates that, in 2023, caregivers to individuals with Alzheimer’s will provide 18 billion hours of care valued at $339.5 billion.
Alzheimer’s Therapeutic Landscape
There are currently several experimental therapeutic agents for Alzheimer’s in various stages of development with clinical testing directed towards amyloid-beta, or Aβ, clearance, and inhibition of Tau protein aggregation or phosphorylated-Tau, or pTau, clearance. In June 2021, the FDA approved Biogen’s Alzheimer’s drug aducanumab, also known as Aduhelm, making it the first medication cleared by U.S. regulators to reduce amyloid plaques in people living with Alzheimer’s and the first new medication for the disease in nearly two decades. There were previously no drugs cleared by the FDA that can slow the mental decline caused by Alzheimer’s, which is the seventh-leading cause of death in the U.S. In July 2023, an anti-beta amyloid antibody known as Lecanemab-irmb (“Leqembi”), received full approval by FDA for treatment of Alzheimer’s. Given the current weight of evidence, amyloid is now established as a cause of Alzheimer’s. Leqembi is a humanized monoclonal antibody that binds with high affinity to soluble amyloid-beta oligomers, which reportedly are toxic to neurons. Leqembi reduced biomarkers of amyloid in early Alzheimer’s and resulted in moderately less decline on measures of cognition and function compared to placebo at 18 months. Since Leqembi only provides passive immunity, antibody infusions are needed every 2 weeks. Leqembi supports and validates the amyloid theory, but in routine medical practice there will be a large burden on the health care system due to the need for every 2-week infusions. As a first-rendition anti-beta amyloid antibody product, it is a major landmark requiring innovation.
| S-10 |
Bipolar Disorder
BD, previously known as manic depression, is a mood disorder characterized by periods of depression and periods of abnormally elevated happiness that each lasts from days to weeks. If the elevated mood is severe or associated with psychosis, it is called mania; if it is less severe, it is called hypomania. During mania, an individual behaves or feels abnormally energetic, happy, or irritable, and they often make impulsive decisions with little regard for the consequences. There is usually also a reduced need for sleep during manic phases. During periods of depression, the individual may experience crying and have a negative outlook on life and poor eye contact with others. The risk of suicide is high; over a period of 20 years, 6% of those with BD died by suicide, while 30–40% engaged in self-harm. Other mental health issues, such as anxiety disorders and substance use disorders, are commonly associated with BD.
While the causes of BD are not clearly understood, both genetic and environmental factors are thought to play a role. Many genes, each with small effects, may contribute to the development of the disorder. Genetic factors account for about 70–90% of the risk of developing BD. Environmental risk factors include a history of childhood abuse and long-term stress. The condition is classified as bipolar I disorder if there has been at least one manic episode, with or without depressive episodes, and as bipolar II disorder if there has been at least one hypomanic episode (but no full manic episodes) and one major depressive episode. If these symptoms are due to drugs or medical problems, they are not diagnosed as BD. Other conditions that have overlapping symptoms with BD include attention deficit hyperactivity disorder, personality disorders, schizophrenia, and substance use disorder as well as many other medical conditions. Medical testing is not required for a diagnosis, though blood tests or medical imaging can rule out other problems.
BD occurs in approximately 1% of the global population. According to the NIH, roughly seven million, are estimated to be affected at some point in their life; rates appear to be similar in females and males. Symptoms most commonly begin between the ages of 20 and 25 years old; an earlier onset in life is associated with a worse prognosis. Interest in functioning in the assessment of patients with BD is growing, with an emphasis on specific domains such as work, education, social life, family, and cognition. Around one-quarter to one-third of people with BD have financial, social or work-related problems due to the illness. BD is among the top 20 causes of disability worldwide and leads to substantial costs for society. Due to lifestyle choices and the side effects of medications, the risk of death from natural causes such as coronary heart disease in people with BD is twice that of the general population.
Bipolar Disorder Therapeutic Landscape
Mood stabilizers, including lithium and certain anticonvulsants, such as valproate and carbamazepine, as well as atypical antipsychotics, such as aripiprazole, are the mainstay of long-term pharmacologic relapse prevention. Antipsychotics are additionally given during acute manic episodes as well as in cases where mood stabilizers are poorly tolerated or ineffective. In patients where compliance is of concern, long-acting injectable formulations are available. There is some evidence that psychotherapy improves the course of BD. The use of antidepressants in depressive episodes is controversial; they can be effective but have been implicated in triggering manic episodes. The treatment of depressive episodes, therefore, is often difficult. Electroconvulsive therapy (“ECT”) is effective in acute manic and depressive episodes, especially with psychosis or catatonia. Admission to a psychiatric hospital may be required if a person is a risk to themselves or others; involuntary treatment is sometimes necessary if the affected person refuses treatment.
Major Depressive Disorder
MDD, also known simply as depression, is a mental disorder characterized by at least two weeks of pervasive low mood, low self-esteem, and loss of interest or pleasure in normally enjoyable activities. Those affected may also occasionally have delusions or hallucinations. Introduced by a group of U.S. clinicians in the mid-1970s, the term was adopted by the American Psychiatric Association for this symptom cluster under mood disorders in the 1980 version of the Diagnostic and Statistical Manual of Mental Disorders (DSM-III) and has become widely used since.
The diagnosis of MDD is based on the person's reported experiences and a mental status examination. There is no laboratory test for the disorder, but testing may be done to rule out physical conditions that can cause similar symptoms. The most common time of onset is in a person’s 20s, with females affected about twice as often as males. The course of the disorder varies widely, from one-episode lasting months to a lifelong disorder with recurrent major depressive episodes.
MDD is believed to be caused by a combination of genetic, environmental, and psychological factors, with about 40% of the risk being genetic. Risk factors include a family history of the condition, major life changes, certain medications, chronic health problems, and substance use disorders. It can negatively affect a person's personal life, work life, or education as well as sleeping, eating habits, and general health. According to the NIH, MDD affected approximately 21 million adults (8.4% of all U.S. adults) in 2020. The prevalence of adults with a major depressive episode was higher among adult females (10.5%) than males (6.2%). The prevalence of adults with a major depressive episode was highest among individuals aged 18-25 (17.0%). MDD causes the second-most years lived with disability, after lower back pain.
| S-11 |
Major Depressive Therapeutic Landscape
Those with MDD are typically treated with psychotherapy and antidepressant medication. Medication appears to be effective, but the effect may predominantly be significant in the most severely depressed. Hospitalization (which may be involuntary) may be necessary in cases with associated self-neglect or a significant risk of harm to self or others. ECT may be considered if other measures are not effective.
Although lithium does not have an FDA approved indication for augmentation of an antidepressant in MDD, it has been prescribed off-label for this purpose for decades. While a wide variety of medications have been used historically in this capacity, lithium is one of the few agents that has demonstrated efficacy in multiple randomized controlled trials. Although the ideal role for lithium augmentation has yet to be established, there is evidence to support the clinical practice of adding lithium to conventional antidepressants in pursuit of MDD remission. Lithium augmentation has been cited as a main strategy for depressed patients not responding to an antidepressant, lithium prophylaxis for recurrent unipolar depression as an alternative to prophylaxis with an antidepressant, and for lithium's anti-suicidal properties, where appropriate.
Post-Traumatic Stress Disorder
PTSD is a mental and behavioral disorder that can develop because of exposure to a traumatic event, such as sexual assault, warfare, traffic collisions, child abuse, domestic violence, or other threats to a person’s life. Symptoms may include disturbing thoughts, feelings, or dreams related to the events, mental or physical distress to trauma-related cues, attempts to avoid trauma-related cues, alterations in the way a person thinks and feels, and an increase in the fight-or-flight response. These symptoms may remain for more than a month after the event. A person with PTSD is at a higher risk of suicide and intentional self-harm.
Most people who experience traumatic events do not develop PTSD. People who experience interpersonal violence such as rape, other sexual assaults, being kidnapped, stalking, physical abuse by an intimate partner, and incest or other forms of childhood sexual abuse are more likely to develop PTSD than those who experience non-assault-based trauma, such as accidents and natural disasters. Those who experience prolonged trauma, such as slavery, concentration camps, or chronic domestic abuse, may develop complex post-traumatic stress disorder (“C-PTSD”). C-PTSD is similar to PTSD but has a distinct effect on a person's emotional regulation and core identity.
According to the NIH, about 3.6%, or roughly nine million, adults in the U.S. have PTSD in a given year, and 9% of people develop it at some point in their life. In much of the rest of the world, rates for a given year are between 0.5% and 1% of the population. Higher rates may occur in regions of armed conflict. It is more common in women than men. PTSD was first mentioned in the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-I) in the 1950s under the term “gross stress reaction.” Although this diagnosis included psychological problems related to traumatic events such as wartime combat, it limited symptoms to six months. This diagnosis was removed from the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-II) in 1968, representing a regression in accurate PTSD characterization. The long-term psychological disabilities experienced by trauma survivors, including Vietnam veterans, sexual assault victims and Holocaust survivors led to the introduction of PTSD in the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-III) in 1980, where, for the first time, the definition of PTSD highlighted the critical connection between traumatic events and long-term psychological symptoms.
Post-Traumatic Stress Disorder Therapeutic Landscape
Prevention may be possible when counselling is targeted at those with early symptoms but is not effective when provided to all trauma-exposed individuals whether or not symptoms are present. The main treatments for people with PTSD are counselling (psychotherapy) and medication. Antidepressants of the selective serotonin reuptake inhibitors (“SSRI”) or serotonin-norepinephrine reuptake inhibitors (“SNRI”) type are the first-line medications used for PTSD and are moderately beneficial for about half of people. Benefits from medication are less than those seen with counselling. It is not known whether using medications and counselling together has greater benefit than either method separately.
Sertraline (Zoloft) and Paroxetine (Paxil) are FDA-approved medications for PTSD. Reviews by a group of doctors of pharmacological monotherapy in 2015 and 2021 found that paroxetine, fluoxetine, sertraline and venlafaxine could be effective for PTSD, but the magnitude of the effect was small and the clinical relevance was unclear. These reviews excluded lithium treatments. Medications, other than some SSRIs or SNRIs, do not have enough evidence to support their use and, in the case of benzodiazepines, may worsen outcomes.
Case reports suggest that lithium treatment may be useful for irritability/anger outbursts in PTSD patients. For example, one study by Kitchner and Greenstein provided case histories of four males (aged approximately 31–42 years) who suffered from PTSD resulting from their experiences in the Vietnam War. Results from treatment with low doses (300–600 mg/day) of lithium carbonate were reported to indicate that treatment was effective in reducing inappropriate anger, irritability, anxiety, and insomnia.
| S-12 |
The clinical observation of mood swings beyond the normal range but milder than those associated with BD reportedly suggested the presence of a subthreshold mood disorder in these PTSD patients. It has also been proposed that treatment of trauma with lithium to forestall the development of PTSD may be provided by pharmacological induction of a mild transient amnesia.
Manufacturing
Currently, we do not have in-house manufacturing capabilities. We have outsourced and expect to continue to outsource the manufacturing of our products to third party contractors, with special capabilities to manufacture chemical drugs and biologic drug candidates for submission and clinical testing under FDA guidelines and, for AL001 and ALZN002, have received Good Manufacturing Practices, or GMP, material manufactured for clinical trial. There are several sources of manufacturing available once a therapy or treatment can achieve Phase II study as identified in a publication by Pharma.org released in 2013 (http://www.phrma.org/sites/default/files/Alzheimer’s%202013.pdf).
Distribution and Marketing
We intend to develop AL001 and ALZN002 through successive de-risking milestones towards regulatory approval and seek marketing approval of AL001 and ALZN002 or enter into partnering transactions with biopharmaceutical companies seeking to strategically fortify pipelines and, in turn, receiving funding for the costly later-stage clinical development required to achieve successful commercialization. We do not anticipate selling products directly into the marketplace, though we may do so depending on market conditions. Our focus is to strategically effect partnering transactions that will provide distribution and marketing capabilities to sell products into the marketplace.
Government Regulation
Clinical trials, the pharmaceutical approval process, and the marketing of pharmaceutical products, are intensively regulated in the United States and in all major foreign countries.
Human Health Product Regulation in the United States
In the United States, the FDA regulates pharmaceuticals under the Federal Food, Drug, and Cosmetic Act and related regulations promulgated thereunder. Pharmaceuticals are also subject to other federal, state, and local statutes and regulations. Failure to comply with applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the imposition by the FDA of an Institutional Review Board, or IRB, a clinical hold on trials, a refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency or judicial enforcement action could have a material adverse effect on us.
The FDA and comparable regulatory agencies in state and local jurisdictions impose substantial requirements upon the clinical development, manufacturing and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, distribution, record keeping, approval, advertising and promotion of our products.
The FDA’s policies may change, and additional government regulations may be promulgated that could prevent or delay regulatory approval of new disease indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or elsewhere.
Marketing Approval
The process required by the FDA before human health care pharmaceuticals may be marketed in the U.S. generally involves the following:
| • | nonclinical laboratory and, at times, animal tests; |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use or uses; |
| • | pre-approval inspection of manufacturing facilities and clinical trial sites; and |
| • | FDA approval of an NDA or BLA, which must occur before a drug or biologic product can be marketed or sold. |
| S-13 |
We will need to successfully complete sufficient clinical trials in order to be in a position to submit a BLA or NDA to the FDA. We must reach agreement with the FDA on the proposed protocols for our future clinical trials in the U.S. A separate submission to the FDA must be made for each successive clinical trial to be conducted during product development. Further, an independent IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that site, and an informed consent must also be obtained from each study subject. Regulatory authorities, a data safety monitoring board or the sponsor may each suspend or terminate a clinical trial at any time on numerous grounds.
For purposes of BLA or NDA approval for human health products, human clinical trials are typically conducted in phases that may overlap.
| • | Phase I. The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase II. This phase involves trials in a limited subject population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. Phase II studies may be sub-categorized into Phase IIA studies which are smaller, pilot studies to evaluate limited drug exposure and efficacy signals, and Phase IIB studies, which are larger studies testing both safety and efficacy more rigorously. |
| • | Phase III. This phase involves trials undertaken to further evaluate dosage, clinical efficacy and safety in an expanded subject population, often at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
All of these trials must be conducted in accordance with Good Clinical Practice (“GCP”), requirements in order for the data to be considered reliable for regulatory purposes.
New Drug and Biologics License Applications
In order to obtain approval to market a pharmaceutical in the United States, a marketing application must be submitted to the FDA that provides data establishing to the FDA’s satisfaction the safety and effectiveness of the investigational drug for the proposed indication. Each NDA or BLA submission requires a substantial user fee payment unless a waiver or exemption applies (such as with the Orphan Drug Designation discussed below). For fiscal year 2023, the FDA set the application fee at $3,242,026 for new drug applications that require clinical data. The manufacturer and/or sponsor of certain drugs approved under an NDA or BLA is also subject to annual prescription drug program fees, currently set at $393,933 per product for fiscal year 2023. These fees are typically increased annually. The NDA or BLA includes all relevant data available from pertinent non-clinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical trials intended to test the safety and effectiveness of the use of a product, or from a number of alternative sources, including studies initiated by investigators.
The FDA will initially review the NDA or BLA for completeness before it accepts it for filing. The FDA has 60 days from its receipt of an NDA or BLA to determine whether the application will be accepted for filing based on the agency’s threshold determination that the application is sufficiently complete to permit substantive review. After the NDA or BLA submission is accepted for filing, the FDA reviews the NDA or BLA to determine, among other things, whether the proposed product is safe and effective for its intended use, and whether the product is being manufactured in accordance with current GMP, or cGMP, to assure and preserve the product’s identity, strength, quality and purity. The FDA may refer applications for novel drug products or drug products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and, if so, under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it typically considers such recommendations carefully when making decisions.
Based on pivotal Phase III trial results submitted in an NDA or BLA, upon the request of an applicant, the FDA may grant a “Priority Review” designation to a product, which sets the target date for FDA action on the application at six to eight months, rather than the standard ten to 12 months. The FDA can extend these reviews by three months. Priority Review is given where preliminary estimates indicate that a product, if approved, has the potential to provide a significant improvement compared to marketed products or offers a therapy where no satisfactory alternative therapy exists. Priority Review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
| S-14 |
After the FDA completes its initial review of an NDA or BLA, it will communicate to the sponsor that the application for the drug will either be approved, or it will issue a complete response letter to communicate that the NDA or BLA will not be approved in its current form and inform the sponsor of changes that must be made or additional clinical, nonclinical or manufacturing data that must be received before the application can be approved, with no implication regarding the ultimate approvability of the application.
Before approving an NDA or BLA, the FDA will inspect the facilities at which the product is manufactured, even if such facilities are located overseas. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications.
Additionally, before approving an NDA or BLA, the FDA may inspect one or more clinical sites and manufacturing sites to assure compliance with GCP and GMP. If the FDA determines that any of the application, manufacturing process or manufacturing facilities is not acceptable, it typically will outline the deficiencies and often will request additional testing or information. This may significantly delay further review of the application. If the FDA finds that a clinical site did not conduct the clinical trial in accordance with GCP, the FDA may determine that the data generated by the clinical site should be excluded from the primary efficacy analyses provided in the NDA or BLA. Additionally, the FDA may identify deficiencies in the manufacturing process and require changes prior to approval. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process for a drug requires substantial time, effort and financial resources, and this process may take up to several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products.
The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called Phase IV studies may be made a condition that must be satisfied for continuing drug approval. The results of Phase IV studies can confirm the effectiveness of a product candidate and can provide important safety information. In addition, the FDA has express statutory authority to require sponsors to conduct post-market studies to specifically address safety issues identified by the agency. Any approvals that we may ultimately receive could be withdrawn if required post-marketing trials or analyses do not meet the FDA requirements, which would materially harm the commercial prospects for AL001 or ALZN002.
The FDA also has authority to require a Risk Evaluation and Mitigation Strategy (“REMS”), from manufacturers to ensure that the benefits of a drug or biological product outweigh its risks. A sponsor may also voluntarily propose a REMS as part of the NDA or BLA submission. The need for a REMS is determined as part of the review of the NDA or BLA. Based on statutory standards, elements of a REMS may include “dear doctor letters,” a medication guide, more elaborate targeted educational programs, and in some cases restrictions on distribution. These elements are negotiated as part of the NDA or BLA approval, and in some cases if consensus is not obtained until after the Prescription Drug User Fee Act review cycle, the approval date may be delayed. Once adopted, a REMS is subject to periodic assessment and modification.
Even if AL001 or ALZN002 receives regulatory approval, the approval may be limited to specific disease states, patient populations and dosages, or might contain significant limitations on use in the form of warnings, precautions or contraindications, or in the form of onerous risk management plans, restrictions on distribution, or post-marketing study requirements. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Any delay in obtaining, or failure to obtain, regulatory approval for AL001 or ALZN002, or obtaining approval only for significantly limited use, would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Breakthrough Therapy Designation
A product can be designated as a breakthrough therapy if it is intended to treat a serious condition (which includes Alzheimer’s) and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). For purposes of breakthrough therapy designation, a clinically significant endpoint generally refers to an endpoint that measures an effect on irreversible morbidity or mortality (“IMM”), or on symptoms that represent serious consequences of the disease. A clinically significant endpoint can also refer to findings that suggest an effect on IMM or serious symptoms, including:
| • | an effect on an established surrogate endpoint; |
| • | an effect on a surrogate endpoint or intermediate clinical endpoint considered reasonably likely to predict a clinical benefit (i.e., the accelerated approval standard); |
| • | an effect on a pharmacodynamic biomarker (which is a measurable indicator of the disease state) that does not meet criteria for an acceptable surrogate endpoint, but strongly suggests the potential for a clinically meaningful effect on the underlying disease; and |
| • | a significantly improved safety profile compared to available therapy (e.g., less dose-limiting toxicity for an oncology agent), with evidence of similar efficacy. |
| S-15 |
A drug that receives a breakthrough therapy designation is eligible for fast-track designation features, intensive guidance on an efficient drug development program and FDA organizational commitment involving senior managers. However, we have not yet applied for breakthrough therapy designation nor have we received any official designation for expedited development. Our product candidates may not qualify for breakthrough therapy designation; further, even if it does qualify for breakthrough therapy designation, it may not actually lead to faster development or expedited regulatory review and approval or necessarily increase the likelihood that it will receive FDA approval.
Based on our preclinical data, AL001 has a positive effect on the pharmacodynamic biomarkers of Alzheimer’s. We propose to validate this clinically and if confirmed, we believe that AL001 is a candidate for breakthrough therapy designation because of its positive effect on a pharmacodynamic biomarker (beta-amyloids) and potential for a clinically meaningful effect on Alzheimer’s. We also believe that ALZN002 is positioned for a breakthrough therapy designation because of its positive effect on a pharmacodynamic biomarker (beta-amyloids) and potential for a clinically meaningful effect on Alzheimer’s.
Section 505(b)(2) New Drug Applications
Companies may also consider seeking FDA approval through the Section 505(b)(2) NDA process if their product candidates are similar to previously approved drugs but differ in dosage form, strength, route of administration, formulation or indication. Section 505(b)(2) of the Food, Drug, and Cosmetic Act was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984 and is also known as the Hatch-Waxman Amendments. The purpose of Section 505(b)(2) is to allow companies to avoid duplicative testing by allowing applicants to utilize data from previous clinical and non-clinical studies in the current NDA submission, when pertinent. The 505(b)(2) application process requires, among other things, the submission of data from studies demonstrating the product’s safety and efficacy for the new indication.
We believe that AL001 is positioned for an expedited Section 505(b)(2) regulatory pathway for a new drug. AL001’s active pharmaceutical ingredients (lithium, proline and salicylate) are well documented and approved by the FDA. The provisions of 505(b)(2) were created, in part, to help avoid unnecessary duplication of studies already performed on a previously approved (“reference” or “listed”) drug. This section gives the FDA express permission to rely on data not developed by the NDA applicant. This process can result in a much less expensive and much faster route to approval, compared with a traditional development path such as 505(b)(1), while creating new, differentiated products with tremendous commercial value.
The Hatch-Waxman Amendments permit companies to rely upon not only certain published nonclinical or clinical studies conducted for an approved product, but also the FDA’s conclusions from a prior review of the studies. Additionally, the FDA may require companies to perform further studies to support changes from the approved product. After completion of the review, the FDA may approve the new product for all or some of the labeled indications for which the reference product has been approved, as well as for any new indication supported by the NDA. While references to nonclinical and clinical data not created by the applicant or for which the applicant does not have a right of reference are allowed, the applicant must still submit data related to the manufacturing and quality of the product candidate, such as information about the development, process, stability, qualification and validation.
If a company chooses to rely on the FDA’s conclusions regarding studies conducted for an already approved product, the company is required to provide a certification statement for any patents listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The FDA will also not approve a Section 505(b)(2) until any non-patent exclusivity period for the reference product has expired, such as the exclusivity granted for obtaining approval of a new chemical entity.
If we qualify for the Section 505(b)(2) regulatory pathway for new drug approvals, we believe we can shorten the development timeline for AL001. However, our AL001 may not qualify for expedited development or, if it does qualify for expedited development, it may not actually lead to faster development or expedited regulatory review and approval.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of certain FDA-regulated products, including prescription drugs, are required to register and disclose certain clinical trial information on a public website maintained by the NIH. Information related to the product, patient population, phase of investigation, study sites and investigator, and other aspects of the clinical trial is made public as part of the registration. Sponsors are also obligated to disclose the results of these trials after completion. Disclosure of the results of these trials can be delayed until the product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the design and progress of our development programs.
| S-16 |
The Drug Price Competition and Patent Term Restoration Act
The Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Amendments, requires pharmaceutical companies to divulge certain information regarding their products, which has the effect of making it easier for other companies to manufacture generic drugs to compete with those products.
Patent Term Extension. After receipt of an NDA or BLA approval, owners of relevant drug patents may apply for a patent extension of up to five years. The permissible patent term extension is calculated as half of the drug’s testing phase, that is, the time between IND submission and NDA or BLA submission, and all of the review phase, or the time between either NDA or BLA submission and approval up to a maximum of five years. The time can be shortened if FDA determines that the applicant did not pursue approval with due diligence. The total patent term after the extension may not exceed 14 years.
For patents that might expire during the application phase, the patent owner may request an interim patent extension. An interim patent extension increases the patent term by one year and may be renewed up to four times. For each interim patent extension granted, the post-approval patent extension is reduced by one year. The director of the U.S. Patent and Trademark Office, or USPTO, must determine that approval of the drug covered by the patent for which a patent extension is being sought is likely. Interim patent extensions are not available for a drug for which an NDA or BLA has not been submitted.
Environmental Regulations. The U.S. generally requires an environmental assessment, which discusses a company’s proposed action, possible alternatives to the action, and whether the further analysis of an environmental impact statement is necessary. Certain exemptions are available from the requirement to perform an environmental assessment and an environmental impact statement. Once an exemption is claimed, a company must state to the FDA that no extraordinary circumstances exist that may significantly affect the environment. We may claim an exemption, under the category for biologic products, from the requirement to provide an environmental assessment and an environmental impact statement for AL001 or ALZN002 and further state to the FDA that, to our knowledge, no extraordinary circumstance exists that would significantly affect the environment.
FDA Post-Approval Requirements
Following the approval of an NDA or BLA, the FDA continues to require adverse event reporting and submission of periodic reports. The FDA also may require post-marketing testing, known as Phase IV testing, REMS, and surveillance to monitor the effects of an approved product, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with FDA and certain state agencies. Registration with the FDA subjects entities to periodic unannounced inspections by the FDA, during which the agency inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMP. Regulatory authorities may withdraw product approvals or request product recalls if a manufacturer fails to comply with regulatory standards, if it encounters problems following initial marketing or if previously unrecognized problems are subsequently discovered.
Patient Protection and Affordable Care Act
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the ACA, which includes measures that have significantly changed the way health care is financed by both governmental and private insurers, became law in the U.S. The ACA is a sweeping measure intended to expand health care coverage within the U.S., primarily through the imposition of health insurance mandates on employers and individuals and expansion of the Medicaid program. The ACA has significantly impacted the pharmaceutical industry. The ACA requires discounts under the Medicare drug benefit program and increased rebates on drugs covered by Medicaid. In addition, the ACA imposes an annual fee, which increases annually, on sales by branded pharmaceutical manufacturers. At this time, the financial impact of these discounts, increased rebates and fees and the other provisions of the ACA on our business are unclear. However, the fees, discounts and other provisions of this law are expected to have a significant negative effect on the profitability of pharmaceuticals.
Human Health Product Regulation in the European Union
In addition to domestic regulations, we may eventually be subject, either directly or through our distribution partners, to a variety of regulations in other jurisdictions governing, among other things, clinical trials and any commercial sales and distribution of our products, if approved.
Whether or not we obtain FDA approval for a product, we must obtain the requisite approvals from regulatory authorities in non-U.S. countries prior to the commencement of clinical trials or marketing of the product in those countries. Certain countries outside of the U.S. have a process that requires the submission of a clinical trial application prior to the commencement of human clinical trials. In Europe, for example, a Clinical Trial Application (“CTA”) must be submitted to the competent national health authority and to independent ethics committees in each country in which a company intends to conduct clinical trials. Once the CTA is approved in accordance with a country’s requirements, clinical trial development may proceed in that country.
| S-17 |
The requirements and process governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country, even though there is already some degree of legal harmonization in the EU Member States resulting from the national implementation of underlying EU legislation. In all cases, the clinical trials are conducted in accordance with GCP and other applicable regulatory requirements.
To obtain regulatory approval of an investigational drug under European Union regulatory systems, we will be required to submit a marketing authorization application. This application is similar to the BLA in the United States, with the exception of, among other things, country-specific document requirements. Drugs can be authorized in the European Union by using (i) the centralized authorization procedure, (ii) the mutual recognition procedure, (iii) the decentralized procedure or (iv) the national authorization procedure.
The European Medicines Agency (“EMA”) implemented the centralized procedure for the approval of human drugs to facilitate marketing authorizations that are valid throughout the EU. This procedure results in a single marketing authorization granted by the European Commission that is valid across the EU, as well as in Iceland, Liechtenstein and Norway, at times referred to as the European Economic Area. The centralized procedure is compulsory for human drugs that: (i) are derived from biotechnology processes, such as genetic engineering; (ii) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative diseases, autoimmune and other immune dysfunctions and viral diseases; (iii) are officially designated orphan drugs; and (iv) constitute advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines. The centralized procedure may at the request of the applicant also be used for human drugs that do not fall within the above mentioned categories if the human drug (a) contains a new active substance which, on the date of entry into force of Regulation (EC) No. 726/2004, was not authorized in the European Economic Area; or (b) the applicant shows that the medicinal product constitutes a significant therapeutic, scientific or technical innovation or that the granting of authorization in the centralized procedure is in the interests of patients at European Economic Area level.
Under the centralized procedure in the European Union, the maximum timeframe for the evaluation of a Marketing Authorization Application by the EMA is 210 days, though the date count stops whenever the Committee for Medicinal Products for Human Use (“CHMP”) asks the applicant for additional written or oral information, with adoption of the actual marketing authorization by the European Commission thereafter. Accelerated evaluation might be granted by the CHMP in exceptional cases, as when a medicinal product is expected to be of a major public health interest from the point of view of therapeutic innovation, defined by three cumulative criteria: (i) the seriousness of the disease to be treated; (ii) the absence of an appropriate alternative therapeutic approach; and (iii) anticipation of exceptional high therapeutic benefit. In this circumstance, EMA ensures that the evaluation for the opinion of the CHMP is completed within 150 days and the opinion issued thereafter.
The Mutual Recognition Procedure (“MRP”) for the approval of human drugs is an alternative approach to facilitate individual national marketing authorizations within the European Union. Essentially, the MRP may be applied for all human drugs for which the centralized procedure is not obligatory. The MRP is applicable to the majority of conventional medicinal products and is based on the principle of recognition of an already existing national marketing authorization by one or more EU Member States.
The principal characteristic of the MRP is that the procedure builds on an already existing marketing authorization in an EU Member State that is used as reference in order to obtain marketing authorizations in other Member States. In the MRP, a marketing authorization for a drug already exists in one or more EU Member States and subsequently marketing authorization applications are made in other EU Member States by referring to the initial marketing authorization. The EU Member State in which the marketing authorization was first granted will then act as the referenced EU Member State. The EU Member States where the marketing authorization is subsequently applied for act as concerned EU Member States.
The MRP is based on the principle of the mutual recognition by EU Member States of their respective national marketing authorizations. Based on a marketing authorization in the reference EU Member State, the applicant may apply for marketing authorizations in other EU Member States. In such case, the reference EU Member State will update its existing assessment report about the drug in 90 days. After the assessment is completed, copies of the report are sent to all EU Member States, together with the approved summary of product characteristics, labeling and package leaflet. The concerned EU Member States then have 90 days to recognize the decision of the referenced EU Member State and the summary of product characteristics, labeling and package leaflet. National marketing authorizations will be granted within 30 days after acknowledgement of the agreement.
If any EU Member State refuses to recognize the marketing authorization by the reference EU Member State on the grounds of potential serious risk to public health, the issue will be referred to a coordination group. Within 60 days, EU Member States will, within the coordination group, make all efforts to reach a consensus. If this fails, the procedure is submitted to an EMA scientific committee for arbitration. The opinion of this EMA Committee is then forwarded to the Commission, for the start of the decision-making process. As in the centralized procedure, this process entails consulting various European Commission Directorates General and the Standing Committee on Human Medicinal Products.
| S-18 |
Human Health Product Regulation in the Rest of World
For countries outside of the EU, such as the United Kingdom, Canada, countries in Eastern Europe or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from country to country. In all cases, the clinical trials are conducted in accordance with GCP and the other applicable regulatory requirements. If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension of clinical trials, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Other Regulatory Considerations
Labeling, Marketing and Promotion. Once an NDA or BLA is approved, or just before approval, a product will be subject to certain marketing and promotional requirements. For instance, the FDA closely regulates the post-approval marketing and promotion of pharmaceuticals, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities on the internet and elsewhere.
While appropriate medical professionals are free to prescribe any pharmaceutical approved by the FDA for any use, a company can only make claims relating to the safety and efficacy of a pharmaceutical that are consistent with the FDA approval, and is only allowed to actively market a pharmaceutical for the particular indication approved by the FDA. Changes to some of the conditions established in an approved application, including changes in indications, labeling, or manufacturing processes or facilities, require submission and FDA approval of a new NDA or BLA or NDA/BLA supplement before the change can be implemented. A BLA supplement for a new indication typically requires clinical data similar to that in the original application, and the FDA uses the same procedures and actions in reviewing supplements as it does in reviewing NDAs.
In addition, any claims we make for our products in advertising or promotion must be appropriately balanced with important safety information and otherwise be adequately substantiated. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising, injunctions and potential civil and criminal penalties. Government regulators recently have increased their scrutiny of the promotion and marketing of pharmaceuticals.
Anti-Kickback and False Claims Laws. In the United States, we are subject to complex laws and regulations pertaining to health care “fraud and abuse,” including, but not limited to, the federal Anti-Kickback Statute, the federal False Claims Act, state false claims acts and anti-kickback statutes, and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular pharmaceutical, for which payment may be made under a federal health care program, such as Medicare or Medicaid.
The federal False Claims Act prohibits anyone from knowingly presenting, or causing to be presented, for payment to federal programs (including Medicare and Medicaid) claims for items or services, including pharmaceuticals, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services.
Many states have similar anti-kickback or false claims statutes that can be even broader than their federal counterparts. There is also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. Many of these laws contain ambiguities as to what is required to comply with the laws. In addition, a federal law known as the Physician Payments Sunshine Act requires pharmaceutical manufacturers to track and report to the federal government certain payments and other transfers of value made to physicians and teaching hospitals and to disclose any physician ownership in the previous calendar year. The data is published annually in a publicly searchable database. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state, and soon federal, authorities.
Other Health Care Laws and Compliance Requirements. In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services (formerly the Health Care Financing Administration), other divisions of the U.S. Department of Health and Human Services (e.g., its Office of Inspector General), the U.S. Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, sales, marketing and scientific/ educational grant programs must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, the privacy provisions of the Health Insurance Portability and Accountability Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veterans Health Care Act of 1992, or VHCA, each as amended, among others. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements will apply. Under the VHCA, drug companies are required to offer certain drugs at a reduced price to a number of federal agencies including U.S. Department of Veteran Affairs and U.S. Department of Defense, the Public Health Service and certain private Public Health Service designated entities in order to participate in other federal funding programs including Medicare and Medicaid. Legislative changes also require that discounted prices be offered for certain U.S. Department of Defense purchases for its TRICARE program via a rebate system. Participation under the VHCA requires submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations.
| S-19 |
In order to distribute products commercially, we must comply with state laws that require the registration of manufacturers and wholesale distributors of pharmaceutical products in a state, including, in certain states, manufacturers and distributors that ship products into the state even if such manufacturers or distributors have no place of business within the state. Some states also impose requirements on manufacturers and distributors to establish the pedigree of product in the chain of distribution, including some states that require manufacturers and others to adopt new technology capable of tracking and tracing product as it moves through the distribution chain. Several states have enacted legislation requiring pharmaceutical companies to establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales, marketing, pricing, clinical trials and other activities or register their sales representatives. Other legislation has been enacted in certain states prohibiting pharmacies and other health care entities from providing certain physician prescribing data to pharmaceutical companies for use in sales and marketing and prohibiting certain other sales and marketing practices. All of our activities are potentially subject to federal and state consumer protection, unfair competition and other laws and regulations.
Our Intellectual Property
We are able to protect our technology from unauthorized use by third parties only to the extent that it is covered by valid and enforceable patents or is effectively maintained as a trade secret or is protected by confidentiality agreements. Accordingly, patents or other proprietary rights are an essential element of our business. Currently, we do not own a patent, although we do possess a license for an immunotherapy technology and three licenses for a lithium, salicylate and proline cocrystal technology from the Licensor.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depending on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
A summary of the licensed patents is as follows:
| Title of Patent | Patent Type | Therapeutic Drug |
Date Filed |
Date Issued |
Expiration Date |
Patent # |
| Lithium Cocrystals and an Additional Neuropsychiatric Agent for Treatment of Neuropsychiatric Disorders |
Method of Use |
AL001 (LISPRO) |
05/21/2016 | 03/28/2017 | 05/21/2036 | 9,603,869 |
| Organic Anion Lithium Ionic Cocrystal Compounds and Compositions | Composition of Matter |
AL001 (LISPRO) |
04/18/2014 | 12/12/2017 | 04/18/2034 | 9,840,521 |
| Amyloid Beta Peptides and Methods of Use | Composition of Matter |
ALZN002 (E22W) |
10/12/2007 | 05/29/2012 | 02/12/2028 | 8,188,046 |
While trade secret protection is an essential element of our business and we take security measures to protect our proprietary information and trade secrets, there can be no assurance that our unpatented proprietary technology will afford us significant commercial protection. We seek to protect our trade secrets by entering into confidentiality agreements with third parties, employees and consultants. However, it is possible that these agreements may be breached or invalidated, and if so, there may not be an adequate corrective remedy available. Accordingly, we cannot ensure that our employees, consultants or any third parties will not breach the confidentiality provisions in our contracts, infringe or misappropriate our trade secrets and other proprietary rights or that measures we take to protect our proprietary rights will be adequate.
In the future, third parties may file claims asserting that our technologies or products infringe on their intellectual property. We cannot predict whether third parties will assert such claims against us or against the licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend ourselves against such claims, whether they are with or without merit and whether they are resolved in favor of, or against, our licensors or ourselves, we may face costly litigation and the diversion of our management’s attention and resources. As a result of such disputes, we may have to develop costly non-infringing technology or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, or at all.
We currently have four trademarks registered with the USPTO that include our corporate name, Alzamend Neuro, two for our corporate slogan and one for our trade name.
| S-20 |
Our Competition
Our industry is highly competitive and subject to rapid and significant technological change. While we have some, albeit limited, development experience and scientific knowledge, we will face competition from both large and small pharmaceutical and biotechnology companies, including specialty pharmaceutical companies and generic drug companies, as well as academic institutions, government agencies and research institutions, among others.
Our competition will be determined in part by the potential indications for which our products are developed and ultimately approved by regulatory authorities. It is likely that the timing of market introductions of some of our potential products or our competitors’ products will be an important competitive factor. Accordingly, the speed with which we can develop our products, conduct preclinical studies and clinical trials to obtain approval and manufacture or obtain supplies of commercial quantities of any approved products should also be important competitive factors. We expect that competition among products approved for sale will be based on additional factors such as product efficacy, safety, reliability, availability, price and patent position.
Employees and Human Capital Resources
As of April 30, 2023, we have four full-time employees and three part-time employees. We also utilize independent consultants to assist us in our medical research and development projects.
Our human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating our existing and new employees, advisors and consultants. The principal purposes of our equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of our company by motivating such individuals to perform to the best of their abilities and achieve our objectives.
Corporate Information
Our principal executive offices are located at 3480 Peachtree Road NE, Second Floor, Suite 103, Atlanta, GA 30326, and our telephone number is (844) 722-6333. Our corporate website address is www.alzamend.com.
| S-21 |
THE OFFERING
The following summary is provided solely for your convenience and is not intended to be complete. You should read the full text and more specific details contained elsewhere in this prospectus. For a more detailed description of our common stock, see “Description of Our Securities.”
| Common stock offered by us pursuant to this prospectus supplement: | Shares of our common stock having an aggregate offering price of up to $9,831,947. | |
| Manner of offering: | “At the market offering” that may be made from time to time through our sales agent, ACM. See “Plan of Distribution” on page S-48. | |
| Use of Proceeds: | We intend to use the net proceeds, if any, from this offering for general corporate purposes, which may include working capital, capital expenditures, research and development expenditures, regulatory affairs expenditures, clinical trial expenditures, acquisitions of new technologies and investments, the financing of possible acquisitions or business expansions, and the repayment, refinancing, redemption or repurchase of future indebtedness or capital stock. See “Use of Proceeds” on page S-47. | |
| Nasdaq Capital Market Symbol | ALZN | |
| Risk Factors: | Investing in our securities is highly speculative and involves a significant degree of risk. See “Risk Factors” and other information included in this prospectus supplement for a discussion of factors you should carefully consider before deciding to invest in our securities. |
| S-22 |
RISK FACTORS
Investing in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should carefully consider the risk factors we describe in any prospectus supplement and in any related free writing prospectus for a specific offering of securities, as well as those incorporated by reference into this prospectus and any prospectus supplement. You should also carefully consider other information contained and incorporated by reference in this prospectus and any applicable prospectus supplement, including our financial statements and the related notes thereto incorporated by reference in this prospectus. The risks and uncertainties described in the applicable prospectus supplement and our other filings with the Commission incorporated by reference herein are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently consider immaterial may also adversely affect us. If any of the described risks occur, our business, financial condition or results of operations could be materially harmed. In such case, the value of our securities could decline and you may lose all or part of your investment.
Risks Related to Our Company, Early Stage of Clinical Development and Financial Condition
We need to obtain substantial additional funding to complete the development and any commercialization of AL001 and ALZN002. If we are unable to raise this capital when needed, we may be forced to delay, reduce or eliminate our research and development programs and other operations.
We expect our expenses to increase substantially during the next few years. The development of biotechnology product candidates is capital intensive. As we conduct non-clinical research and clinical development of our product candidates, we will need substantial additional funds to maintain and expand our capabilities in a variety of areas including discovery and non-clinical research, clinical development, regulatory affairs, product development, product quality assurance, and pharmacovigilance. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses for marketing, sales, manufacturing and distribution. Some of those commercialization investments may be made at-risk in advance of receiving an approval.
As of April 30, 2023, we had $5.1 million in cash and cash equivalents. Based on our current operating plan, we believe that without additional funding, our existing cash and cash equivalents will enable us to fund our operations for approximately six months. In particular, we need additional funds to allow us to fund Phase II clinical trials for AL001 in Alzheimer’s, BD, MDD and PTSD and to complete the on-going phase I/IIA clinical trial for ALZN002 to treat mild to moderate dementia of the Alzheimer’s type. However, changing circumstances or inaccurate estimates by us may cause us to use capital significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. For example, our ongoing clinical trial for ALZN002 or our planned clinical trials for AL001 may encounter technical, enrollment or other issues that could cause our development costs to increase more than we expect. We will not have sufficient funds to complete any of these planned or ongoing clinical trials or the clinical development of either AL001 or ALZN002 through regulatory approval. We will need to raise substantial additional capital to complete the development and commercialization of each of those product candidates, which additional capital, if available on reasonable terms if at all, may be raised through the sale of our common stock or other securities or through the entering into of alternative strategic transactions, or cause our stockholders to incur substantial dilution.
Our future capital requirements will depend on many factors, including:
| • | the initiation, progress, timing, costs and results of our planned clinical trials for our product candidates; |
| • | the number and scope of indications we decide to pursue for product development; |
| • | the cost, timing and outcome of regulatory review of any NDA or BLA we may submit for our product candidates; |
| • | the costs and timing of manufacturing for our product candidates, if approved; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| • | our efforts to enhance operational systems and our ability to attract, hire and retain qualified personnel, including personnel to support the development of our product candidates; |
| • | the costs associated with being a public company; |
| • | our ability to enter into partnerships or otherwise monetize our pipeline through strategic transactions on a timely basis, on terms that are favorable to us, or at all; |
| • | the terms and timing of establishing and maintaining collaborations, licenses and other similar arrangements; |
| S-23 |
| • | the extent to which we acquire or in-license other product candidates and technologies; and |
| • | the cost associated with commercializing our product candidates, if any are approved for commercial sale. |
Our commercial revenues, if any, will be derived from sales of products that we do not expect to be commercially available for sale for at least the next several years, if ever. Accordingly, we will need to obtain substantial additional funding in connection with our continuing operations. Adequate additional financing may not be available to us on acceptable terms, or at all. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. If we are unable to raise capital when needed or on attractive terms, we would be forced to delay, reduce or eliminate our research and development programs or other operations.
Our independent registered public accounting firm has expressed substantial doubt about our ability to continue as a going concern.
Our auditors have issued a going concern opinion on our financial statements for the year ended April 30, 2023, expressing substantial doubt that we can continue as an ongoing business due to insufficient capital for us to fund our operations. Our financial statements do not include any adjustments that may result from the outcome of this uncertainty. If we are unable to successfully raise additional capital, we will need to create and implement alternate operational plans to continue as a going concern, and investors or other financing sources may be unwilling to provide additional funding to us on commercially reasonable terms or at all.
We are at an early stage of clinical development and currently have no source of near-term revenue and may never become profitable.
We are a clinical-stage biopharmaceutical company. We have recently initiated clinical trials for our AL001 and ALZN002 programs. To date, we have not initiated or completed a pivotal clinical trial, obtained marketing approval for any product candidates, manufactured a commercial scale product or arranged for a third party to do so on our behalf, or conducted sales and marketing activities necessary for successful product commercialization. Our ability to generate revenue depends heavily on, among other developments:
| • | demonstration to the satisfaction of the FDA and comparable regulatory bodies that AL001 and ALZN002 are safe and effective in future clinical trials; |
| • | our ability to seek and obtain regulatory approvals, including with respect to the indications we are seeking; |
| • | if approved by the FDA, successful manufacture and commercialization of AL001 and ALZN002; and |
| • | market acceptance of AL001 and ALZN002. |
We only have two product candidates, AL001 and ALZN002, which will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before either or both of them, and any respective successors, will provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval for and commercialize AL001 or ALZN002, we will be unable to generate any revenue for many years, if at all. We do not anticipate that we will generate revenue for a few years, at the earliest, or that we will achieve profitability for at least several years after generating material revenue, if at all. If we are unable to generate revenue, we will not become profitable, and we may be unable to continue our operations.
We have a limited operating history on which to judge our business prospects and management.
We were incorporated in February 2016 and commenced operations shortly thereafter. We have a limited operating history upon which to base an evaluation of our business and prospects. Operating results for future periods are subject to numerous uncertainties and we cannot assure you that we will achieve or sustain profitability. Our prospects must be considered in light of the risks encountered by companies in the early stage of development, particularly companies in new and rapidly evolving markets. Future operating results will depend upon many factors, including our success in attracting and retaining motivated and qualified personnel, our ability to establish short term credit lines or obtain financing from other sources, our ability to develop and market new products or control costs, and general economic conditions. We cannot assure you that we will successfully address any of these contingencies.
| S-24 |
Risks Related to Our Product Candidates
We have both operational and financial milestones that must be met to maintain the licensing rights to our current technology and intellectual property from the University of South Florida Research Foundation.
There are certain license fees and milestone payments required to be paid by us to the Licensor, pursuant to the terms of license agreements we have entered into with the Licensor. The license agreements for ALZN002 require us to pay royalty payments of 4% on net sales of products developed from the licensed technology for ALZN002 while the license agreements for AL001 require that we pay combined royalty payments of 4.5% on net sales of products developed from the licensed technology for AL001. We have already paid an initial license fee of $200,000 for ALZN002 and an initial license fee of $200,000 for AL001. As an additional licensing fee for the license of ALZN002, the Licensor received 3,601,809 shares of our common stock. As an additional licensing fee for the license of the AL001 technologies, the Licensor received 2,227,923 shares of our common stock. Minimum royalties for AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the AL001 License Agreements. Minimum royalties for ALZN002 are $20,000 on the first anniversary of the first commercial sale, $40,000 on the second anniversary first commercial sale and $50,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the ALZN002 License Agreement. Minimum royalties for November AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the November AL001 License Agreements ..Additionally, we are required to pay milestone payments on the due dates to the Licensor for the license of the AL001 technologies and for the ALZN002 technology, as follows:
Original AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Completed September 2019 | Pre-IND meeting | |
| $ | 65,000 | * | Completed June 2021 | IND application filing | |
| $ | 190,000 | * | Completed December 2021 | Upon first dosing of patient in a clinical trial | |
| $ | 500,000 | * | Completed March 2022 | Upon Completion of first clinical trial | |
| $ | 1,250,000 | 24 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 8 years from the effective date of the agreement | Upon FDA NDA approval | ||
| * | Milestone met and completed |
We have met the pre-IND meeting, IND application filing, and successfully completed the Phase I clinical trial milestones encompassing AL001. If we fail to meet a milestone payment by the specified date, the Licensor may terminate the respective license agreement. If the Licensor were to terminate either license agreement for whatever reason, it would materially and adversely affect our business, financial position and future prospects and you would likely lose the entirety of your investment in us.
ALZN002 License:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Completed January 2022 | Upon IND application filing | |
| $ | 50,000 | September 2023 | Upon first dosing of patient in first Phase I clinical trial | ||
| $ | 500,000 | 24 months from completion of first Phase I clinical trial | Upon completion of first Phase II clinical trial | ||
| $ | 1,000,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA BLA approval | ||
| * | Milestone met and completed |
Additional AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 2,000,000 | 36 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 16,000,000 | August 1, 2029 | First commercial sale | ||
These June AL001 License Agreements have an indefinite term that continue until the later of the date no licensed patent under the applicable agreement remains a pending application or enforceable patent, the end date of any period of market exclusivity granted by a governmental regulatory body, or the date on which the licensee’s obligations to pay royalties expire under the applicable license agreement.
| S-25 |
If we fail to comply with our obligations in the agreements under which we license intellectual property and other rights from third parties or otherwise experience disruptions to our business relationships with the Licensor, we could lose license rights that are important to our business.
We are a party to these license agreements with the Licensor and expect to enter into additional license agreements in the future. The existing license agreements impose, and we expect that future license agreements will impose, various diligence, milestone payment, royalty and other obligations on us. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, we may be required to make certain payments to the Licensor, we may lose the exclusivity of our license, or the Licensor may have the right to terminate the license, in which event we would not be able to develop or market products covered by the license. The Licensor or any future licensor may take any of these actions, including terminating a license agreement. Additionally, the milestone and other payments associated with these licenses will make it less profitable for us to develop our product candidates. If the Licensor were to terminate a license agreement for whatever reason, it would materially and adversely affect our business, financial position and future prospects and you would likely lose the entirety of your investment in us.
In some cases, patent prosecution of our licensed technology is controlled solely by the Licensor. If the Licensor fails to obtain and maintain patent or other protection for the proprietary intellectual property we license, we could lose our rights to the intellectual property or our exclusivity with respect to those rights, and our competitors could market competing products using the intellectual property. Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise regarding intellectual property subject to a licensing agreement, including but not limited to:
| • | the scope of rights granted under the license agreement and other interpretation-related issues; |
| • | the extent to which our technology and processes infringe on intellectual property of the Licensor that is not subject to the licensing agreement; |
| • | the sublicensing of patent and other rights; |
| • | our diligence obligations under each of the license agreements and what activities satisfy those diligence obligations; |
| • | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our collaborators; and |
| • | the priority of invention of patented technology. |
If disputes over intellectual property and other rights that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
We are substantially dependent on the success of our product candidates, which may not receive regulatory approval or be successfully commercialized.
In the future, we plan to submit AL001 and ALZN002 and, potentially, other product candidates for regulatory approval. Currently, however, neither AL001 nor ALZN002 has been submitted for regulatory approval, which would be required before we seek to initiate commercial distribution. To date, we have invested nearly all of our resources in establishing our company and the acquisition of the intellectual property of our product candidates, AL001 and ALZN002. Our near-term prospects, including our ability to finance our company and to enter into strategic collaborations and, ultimately, to generate revenue, are directly dependent upon the successful development, FDA approval and commercialization of AL001 or ALZN002.
The development and commercial success of our product will depend on a number of factors, including, without limitation, the following:
| • | our timely initiation and successful completion of preclinical studies and clinical trials for AL001 or ALZN002; |
| • | our demonstration to the satisfaction of the FDA and comparable regulatory bodies of the safety and efficacy of AL001 or ALZN002, as well as to obtain regulatory and marketing approval for AL001 or ALZN002 in the United States, Europe, the United Kingdom and elsewhere; |
| • | our continued compliance with all clinical and regulatory requirements applicable to AL001 and ALZN002; |
| • | our maintenance of an acceptable safety profile of AL001 and ALZN002 following regulatory approval; |
| S-26 |
| • | competition with other treatments; |
| • | our creation, maintenance and protection of our intellectual property portfolio, including patents and trade secrets, and regulatory exclusivity for AL001 and ALZN002; |
| • | the effectiveness of our and our eventual partners’ marketing, sales and distribution strategy and operations; |
| • | the ability of our third-party manufacturers to manufacture supplies of our product and product candidates and to develop, validate and maintain commercially viable manufacturing processes; |
| • | our ability to launch commercial sales of AL001 or ALZN002 following regulatory approval, whether alone or in collaboration with others; and |
| • | the acceptance of AL001 and ALZN002 by physicians, health care payers, patients and the medical community. |
Many of these factors are beyond our control, and we cannot assure you that we will ever be able to generate sufficient revenue, or any revenue at all, from the sale of AL001 or ALZN002. Our failure in any of the above factors, or in successfully commercializing AL001 or ALZN002 on a timely basis, could have a material adverse effect on our business, results of operations and financial condition, and the value of your investment could substantially decline.
AL001 and ALZN002 may not achieve market acceptance, which would significantly limit our ability to generate revenue.
Even if we develop AL001 or ALZN002 and gain regulatory approvals for either or both candidates, unless physicians and patients accept our product candidates, we may not be able to sell them, whether directly or indirectly, and generate significant revenues. We cannot assure you that AL001, ALZN002 or any other potential product candidates we may eventually develop will achieve market acceptance and revenue if and when they obtain the requisite regulatory approvals. Market acceptance of any product candidate depends on a number of factors, including but not limited to:
| • | the indication and warnings approved by regulatory authorities in the product label; |
| • | continued demonstration to the FDA of safety and efficacy in commercial use; |
| • | physicians’ willingness to prescribe the product; |
| • | reimbursement from third-party payers such as government health care systems and insurance companies; |
| • | the price of the product; |
| • | the nature of any post-approval risk management plans mandated by regulatory authorities; |
| • | competition; and |
| • | the effectiveness of marketing and distribution support. |
Any failure by AL001 or ALZN002 to achieve market acceptance or commercial success could have a material adverse effect on our business, results of operations and financial condition.
Problems in the manufacturing process, failure to comply with manufacturing regulations or unexpected increases in manufacturing costs could harm our business, results of operations and financial condition.
We are responsible for the manufacture and supply of AL001 and ALZN002 independently of each other. The manufacturing of AL001 and ALZN002 necessitates compliance with applicable regulatory requirements of the FDA and the European Union, as well as with international cGMP and other international regulatory requirements. As of the date of this Annual Report, we do not have our own manufacturing facilities. We have contracted with a third-party manufacturer for the clinical supply of AL001 using GMP manufacturing for our planned AL001 clinical trials and plan to contract with established third parties for the long-term commercial production of AL001 and ALZN002. The responsibility to obtain market authorization for AL001 and ALZN002 remains with us. As such, even if we could potentially have a claim against one or more third parties, we are legally liable for any noncompliance related to AL001 and ALZN002 and we expect to retain legal responsibility for any future product candidates as well.
Additionally, we may have limited control over the associated manufacturing costs and potential unexpected increases in those costs over time. If costs increase, we may choose to pass on such costs to our customers, which could reduce our ability to compete by increasing the prices of our products (which we expect to be priced at a significant premium over competing generic products). See “Risks Related to Our Business and Industry — We expect to face substantial competition, with other entities possibly discovering, developing or commercializing products before, or more successfully than, we do.” If we cannot pass on all such costs to our customers, then our profitability would be adversely affected.
| S-27 |
If we are unable to manufacture, or contract to manufacture, AL001 and ALZN002 in accordance with regulatory specifications, or if there are disruptions in the manufacturing process due to damage, loss or failure to meet regulatory requirements (including passing inspections) of manufacturing facilities, we may not be able to meet the demand for our products or supply sufficient product for use in clinical trials, and this may harm our ability to commercialize AL001 and ALZN002 on a timely or cost-competitive basis, or preclude us from doing so at all, which could harm our business, results of operations and financial condition.
Before we or any future commercial partners can begin commercial manufacture of AL001 and ALZN002 or any other product candidate that we may develop in the future, we must obtain FDA regulatory approval for the product, which requires a successful FDA inspection of our manufacturing facilities (or those we contract with) and the development of quality systems, among other requirements. Even if we successfully pass an FDA Pre-Approval Inspection of any manufacturing facilities we may establish or contract with, our pharmaceutical facilities would be subject to unannounced inspection by the FDA and foreign regulatory authorities to ensure ongoing manufacturing compliance, even after product approval. Due to the complexity of the processes that we anticipate will eventually be used to manufacture AL001 and ALZN002, we may be unable to pass federal, state or international regulatory inspections in a cost-effective manner, whether initially or at any time thereafter. If we are unable to comply with manufacturing regulations, we may be subject to fines, unanticipated compliance expenses, recall or seizure of any approved products, or legal actions such as injunctions or criminal or civil prosecution. These possible sanctions could materially and adversely affect our business, results of operations and financial condition. See also “Risks Related to Development and Regulatory Approval of Our Product.” The regulatory approval process is uncertain, requires us to utilize significant financial, physical and human resources, and may prevent us or our future commercial partners from obtaining approvals for the commercialization of some or all of our product candidates.
Serious adverse events or other safety risks could require us to abandon development and preclude, delay or limit approval of AL001 or ALZN002, or limit the scope of any approved label or market acceptance.
If AL001, ALZN002 or any other product candidate that we may develop in the future, prior to or after any approval for commercial sale, causes serious or unexpected side effects, or become associated with other safety risks such as misuse, abuse or diversion, a number of potentially significant negative consequences could result, including, without limitation, that:
| • | regulatory authorities may interrupt, delay or halt clinical trials; |
| • | regulatory authorities may deny regulatory approval of AL001 or ALZN002; |
| • | regulatory authorities may require certain labeling statements, such as warnings or contraindications or limitations on the indications for use, or impose restrictions on distribution in the form of REMS in connection with approval, if any; |
| • | regulatory authorities may withdraw their approval, require more onerous labeling statements or impose a more restrictive REMS of any product that is approved; |
| • | we may be required to change the way the product is administered or conduct additional clinical trials; |
| • | any relationships that we may be able to form in the future with any commercial partners may suffer; |
| • | we could be sued and held liable for harm caused to patients; and |
| • | our reputation may suffer. |
We may voluntarily suspend or terminate our clinical trials if at any time we believe that they present an unacceptable risk to participants or if preliminary data demonstrate that either AL001 or ALZN002 is unlikely to receive regulatory approval or is unlikely to be successfully commercialized. In addition, regulatory agencies, an Ethics Committee or Institutional Review Board (an “IRB”), or data safety monitoring boards may at any time recommend the temporary or permanent discontinuation of our clinical trials or request that we cease using investigators in the clinical trials if they believe that the clinical trials are not being conducted in accordance with applicable regulatory requirements, or that they present an unacceptable safety risk to participants. If we elect or are forced to suspend or terminate a clinical trial of AL001, ALZN002 or any other product candidate that we may in the future develop, the commercial prospects for that product will be harmed and our ability to generate product revenue from that product may be delayed or eliminated. Furthermore, any of these events could prevent us or our partners from achieving or maintaining market acceptance of the affected product and could substantially increase the costs of commercializing AL001 or ALZN002 and materially impair our ability to generate revenue from the commercialization of AL001 or ALZN002 either by us or by any future commercial partners with which we may develop a relationship, which and could have a material adverse effect on our reputation, business, results of operations and financial condition.
| S-28 |
If we fail to obtain and sustain an adequate level of reimbursement for our products by third-party payers, sales and profitability will be adversely affected.
The course of medical treatment for human patients is, and will continue to be, expensive. We expect that most patients and their families will not be capable of paying for our potential products themselves.
Accordingly, it is unlikely that there will be a commercially viable market for AL001 or ALZN002, if approved, without reimbursement and coverage from third-party payers. Obtaining reimbursement approval and coverage from third-party payers is a time consuming and expensive process, and we cannot be certain that reimbursement will be approved and coverage obtained for our current product candidates or any other product candidate we may develop. Additionally, even if there is some form of reimbursement and coverage from third-party payers, if the level of third-party reimbursement is insufficient from the patient’s perspective or coverage is limited, our revenue and gross margins will be materially and adversely affected.
A current trend in the U.S. health care industry, as well as in other countries around the world, is toward cost containment. Large public and private payers, managed care organizations, group purchasing organizations and similar organizations are exerting increasing influence on decisions regarding the use of, and reimbursement levels for, particular treatments. Third-party payers, such as government programs, including Medicare in the United States, and private health care insurers, carefully review and have increasingly been challenging the coverage of, and prices charged for, medical products and services. Many third-party payers limit coverage of or reimbursement for newly-approved health care products. Reimbursement rates and coverage from private health insurance companies vary depending on the company, the insurance plan and other factors. Cost-control initiatives could decrease the price we or our partners establish for products, which could result in lower product revenue and profitability.
Reimbursement systems in international markets vary significantly by country and by region, and reimbursement approvals must be obtained on a country-by-country basis. Our eventual partners may elect to reduce the price of our products in order to increase the likelihood of obtaining reimbursement approvals. In many countries, products cannot be commercially launched until reimbursement is approved and the negotiation process in some countries can exceed 12 months. In addition, pricing and reimbursement decisions in certain countries can be affected by decisions taken in other countries, which can lead to mandatory price reductions and/or additional reimbursement restrictions across a number of other countries, which may adversely affect our sales and profitability. If countries set prices that are not sufficient to allow us or our partners to generate a profit, our partners may refuse to launch the product in such countries or withdraw the product from the market, which would adversely affect our sales and profitability and could materially and adversely affect our business, results of operations and financial condition.
Risks Related to Development and Regulatory Approval of Our Drug Candidates
The regulatory approval process is uncertain, requires us to utilize significant resources, and may prevent us or our future commercial partners from obtaining approvals for the commercialization of AL001 or ALZN002.
The research, testing, manufacturing, labeling, approval, sale, marketing and testing of AL001 and ALZN002 are and will be subject to extensive regulation by regulatory authorities in the United States, Europe and elsewhere, and regulatory requirements applicable to our product differ from country to country. Neither we nor any commercial partner will be permitted to market any of our current or future product candidates in the United States until we receive approval from the FDA of either a NDA or BLA for AL001 and ALZN002, respectively. Obtaining approval of an NDA or a BLA is an uncertain process that requires us to utilize significant resources. Furthermore, regulatory authorities possess broad discretion regarding processing time and usually request additional information and raise questions which have to be answered. There is considerable uncertainty regarding the times at which products may be approved and we have no control over the FDA review process. In addition, failure to comply with FDA and other applicable U.S. and foreign regulatory requirements may subject us to administrative or judicially imposed sanctions, including: warning letters, civil and criminal penalties, injunctions, withdrawal of approved products from the market, product seizure or detention, product recalls, total or partial suspension of production, and refusal to approve pending applications or supplements to approved applications.
Even if we fully comply with all applicable laws and regulations, the FDA may still determine that our clinical data are insufficient for final approval of an NDA or BLA. The process required by the FDA and most foreign regulatory authorities before human health care pharmaceuticals may be marketed generally involves nonclinical laboratory and, in some cases, animal tests; submission of an IND, which must become effective before clinical trials may begin; adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use or uses; pre-approval inspection of manufacturing facilities and clinical trial sites; and FDA approval of an NDA or BLA, which must occur before a drug can be marketed or sold.
Regulatory approval of an NDA or BLA, or any supplement thereof, is not guaranteed, and the approval process requires us to utilize significant resources, could take several years, and is subject to the substantial discretion of the FDA. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon or have to repeat or perform additional studies. If our product or any of our future product candidates fails to demonstrate safety and efficacy in our studies, or for any other reason does not gain regulatory approval, our business and results of operations will be materially and adversely harmed.
| S-29 |
In addition, separate regulatory approvals are required in order to market any product in many jurisdictions, including the United States, the United Kingdom, European Economic Area, which consists of the 27 Member States (known as the “EU Member States”) of the European Union plus Norway, Iceland and Liechtenstein, and others. Approval procedures vary among countries and can involve additional studies and testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Studies conducted in one country may not be accepted by regulatory authorities in other countries. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one or more foreign regulatory authorities does not ensure approval by regulatory authorities in other foreign countries or by the FDA. However, a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval. We may be unable to file for regulatory approvals or do so on a timely basis and, even if we are able to, we may not receive necessary approvals to commercialize our products in any market. Any of these results could have a material adverse effect on our business, results of operations and financial condition.
There is a high rate of failure for drug candidates proceeding through clinical trials.
Generally speaking, there is a high rate of failure for drug candidates proceeding through clinical trials. We may suffer significant setbacks in our clinical trials similar to the experience of a number of other companies in the pharmaceutical and biotechnology industries, even after receiving promising results in earlier trials. Further, even if we view the results of a clinical trial to be positive, the FDA or other regulatory authorities may disagree with our interpretation of the data. For instance, any such differing interpretation could cause the FDA to require additional trials. In the event that:
| (i) | we obtain negative or inconclusive results from the AL001 or ALZN002 from a clinical trial; |
| (ii) | the FDA places a clinical hold on our clinical trials due to potential chemistry, manufacturing and controls issues or other hurdles; or |
| (iii) | the FDA does not approve our NDA for AL001 or our BLA for ALZN002, then: |
| • | we may not be able to generate sufficient revenue or obtain financing to continue our operations; |
| • | our ability to execute our current business plan will be materially impaired; |
| • | our reputation in the industry and in the investment community would likely be significantly damaged; and |
| • | the price of our common stock would likely decrease significantly. |
Any of these results could materially and adversely affect our business, results of operations or financial condition.
Nearly every attempt at drug approval for Alzheimer’s has failed.
Despite billions of dollars invested by the NIH and the biopharmaceutical industry in research programs to develop novel therapeutics for Alzheimer’s, the FDA has only approved two new drugs for Alzheimer’s since 2003; in June 2021, aducanumab (Biogen, Inc) received approval from the FDA for the treatment of Alzheimer’s using the accelerated approval pathway and in July 2023, Leqembi (Eisai) received full approval by the FDA for treatment of Alzheimer’s disease. Since 2003, many new types and classes of drugs have been developed and tested in Alzheimer’s, including monoclonal antibodies, gamma secretase modulators and inhibitors, β-site amyloid precursor protein cleaving enzyme inhibitors, receptor for advanced glycation end-products inhibitors, nicotinic partial agonists and allosteric modulators, serotonin subtype receptor antagonists, and others. Except for Biogen’s and Eisai’s approvals, referred to above, virtually all of these scientific programs have failed in clinical testing.
Clinical trials for AL001 or ALZN002 can be expensive, time consuming, uncertain and susceptible to change, delay or termination.
Clinical trials are expensive, time consuming and difficult to design and implement. The result of a clinical trial may be undesirable and can result in a clinical trial cancellation or the need for re-evaluation and supplementation. Even if the results of our clinical trials are favorable, the clinical trials for AL001 or ALZN002 are expected to continue for a few years and may even take significantly longer to complete. In addition, we, the FDA, an IRB, or other regulatory authority, whether in the United States, European Union or elsewhere, may suspend, delay or terminate our clinical trials at any time, for various reasons, including, without limitation:
| • | lack of effectiveness of AL001 or ALZN002 during clinical trials; |
| • | discovery of serious or unexpected toxicities or side effects experienced by trial participants or other safety issues; |
| • | slower than expected rates of subject recruitment and enrollment rates in clinical trials; |
| S-30 |
| • | difficulty in retaining subjects who have initiated a clinical trial but may have withdrawn due to adverse side effects from the therapy, insufficient efficacy, fatigue with the clinical trial process or for any other reason; |
| • | delays or inability in manufacturing or obtaining sufficient quantities of materials for use in clinical trials due to manufacturing or regulatory constraints; |
| • | inadequacy of or changes in our manufacturing process or product formulation; |
| • | delays in obtaining regulatory authorization to commence a trial, including experiencing “clinical holds” or delays requiring suspension or termination of a trial by a regulatory agency, such as the FDA, before or after a trial is commenced; |
| • | changes in applicable regulatory policies and regulations; |
| • | delays or failure in reaching agreement on acceptable terms in clinical trial contracts or protocols with prospective clinical trial sites; |
| • | delay or failure to supply product for use in clinical trials which conforms to regulatory specification; |
| • | unfavorable results from ongoing preclinical studies and clinical trials; |
| • | failure of any contract research organizations (“CROs”) that we may partner with in the future, or other third-party contractors, to comply with all contractual requirements or to perform their services in a timely or acceptable manner; |
| • | failure by us, our employees, any CROs or their employees to comply with all applicable FDA or other regulatory requirements relating to the conduct of clinical trials; |
| • | scheduling conflicts with participating clinicians and clinical institutions; |
| • | failure to design appropriate clinical trial protocols; or |
| • | regulatory concerns with pharmaceutical products generally and the potential for abuse. |
The occurrence of any of the foregoing could have a material adverse effect on our business, results of operations and financial condition. See the risk factor “There is a high rate of failure for drug candidates proceeding through clinical trials” above.
If our products do not receive breakthrough therapy designation, it could potentially increase the FDA’s review time and adversely impact our development timeline. Even if the FDA grants breakthrough therapy designation, it does not guarantee faster product development or FDA review and does not necessarily increase the likelihood of the product candidates receiving approval from the FDA.
Breakthrough therapy designation is reserved for drug or biologic products that are intended to treat serious conditions and for which preliminary clinical evidence indicates that the candidate may demonstrate a substantial improvement on one or more clinically significant endpoints over currently available therapies. The benefits of receiving the designation include additional guidance from FDA throughout the development process, assistance with designing clinical trials, and coordination with FDA senior managers and experienced review staff. We plan to seek breakthrough therapy designation for both AL001 and ALZN002. However, we have neither received breakthrough therapy designation nor have we qualified for expedited development, and no assurance can be given that we will. Even if we qualify for breakthrough therapy designation or expedited development, it may not actually lead to faster development or expedited regulatory review and approval or necessarily increase the likelihood that we will receive FDA approval.
Even if we believe that our products are strong candidates for breakthrough therapy designation, it is possible that the FDA may determine that our preliminary clinical evidence is insufficient to justify breakthrough therapy designation. Without this designation, we would not be able to benefit from the increased FDA guidance and assistance throughout the development process, and it is possible that our development timeline could be extended.
The breakthrough therapy designation, while at times advantageous for the development process for the reasons identified above, may nevertheless have little or no positive impact on our development process. There is no guarantee that, even with the FDA’s assistance through the breakthrough therapy designation, that the development process will be accelerated, the FDA will review or approve our submissions in a timely manner, or that our product candidates will ultimately receive approval from the FDA.
In summary, we cannot guarantee that our product candidates will receive breakthrough therapy designations and, even if one does, we cannot guarantee that such designations will have any bearing on the FDA’s review or approval of our product candidates.
| S-31 |
Even if we receive regulatory approval for any of our future product candidates, we will be subject to ongoing FDA and other regulatory body obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, will be subject to labeling and manufacturing requirements and could be subject to other restrictions. Failure to comply with these regulatory requirements or the occurrence of unanticipated problems with our products could result in significant penalties.
Any regulatory approvals that we or any of our collaborators receive for AL001, ALZN002 or any future product candidate may be subject to conditions of approval or limitations on the approved indicated uses for which the product may be marketed or may contain requirements for potentially costly surveillance to monitor the safety and efficacy of the product candidate. In addition, AL001, ALZN002 and any of our future product candidates, if approved by the FDA or other regulatory bodies, will be subject to extensive and ongoing regulatory requirements regarding the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping. These requirements will include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMP, Good Laboratory Practice and Good Clinical Practice, the three types of audits related to the progressive stages needed to bring a pharmaceutical product to market, for any studies that we conduct post-approval. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| • | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| • | fines, warning letters or holds on target studies; |
| • | refusal by the FDA or other applicable regulatory body to approve pending applications or supplements to approved applications filed by us or our strategic collaborators, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; and |
| • | injunctions or the imposition of civil or criminal penalties. |
The policies of the FDA and other regulatory bodies may change, and additional government regulations may be promulgated that could prevent, limit or delay regulatory approval of AL001 or ALZN002. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or elsewhere. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, and we may not achieve or sustain profitability, which would materially and adversely affect our business, results of operations and financial condition.
AL001 or ALZN002 and any of our future product candidates, if approved, may cause or contribute to adverse medical events that we are required to report to the FDA and regulatory authorities in other countries and, if we fail to do so, we could be subject to sanctions that would materially harm our business.
If we are successful in commercializing AL001, ALZN002 or any of our future product candidates, regulations promulgated by the FDA and by the regulatory authorities in other countries require that we report certain information about adverse medical events if those products may have caused or contributed to those adverse events. The timing of our obligation to report would be triggered by the date we become aware of the adverse event as well as the nature of the event. We may fail to report adverse events we become aware of within the prescribed timeframe. We may also fail to appreciate that we have become aware of a reportable adverse event, especially if it is not reported to us as an adverse event or if it is an adverse event that is unexpected or removed in time from the use of our products. If we fail to comply with our reporting obligations, the FDA and regulatory authorities in other countries could take action including criminal prosecution, the imposition of civil monetary penalties, seizure of our products, or delay in approval or clearance of future products, which could have a material adverse effect on our business, results of operations and financial condition.
Legislative or regulatory reforms with respect to products may make it more difficult and costly for us to obtain regulatory clearance or approval of AL001, ALZN002 or any of our future product candidates and to produce, market, and distribute our products after clearance or approval is obtained.
From time to time, legislation is drafted and introduced in the U.S. Congress and lawmaking bodies in other countries that could significantly change the statutory provisions governing the testing, regulatory clearance or approval, manufacture, and marketing of regulated products. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Similar changes in regulations can occur in other countries. Any new regulations or revisions or reinterpretations of existing regulations in the United States or in other countries may impose additional costs or lengthen review times of AL001, ALZN002 and any of our future product candidates. We cannot determine what effect changes in regulations, statutes, legal interpretation or policies, when and if promulgated, enacted or adopted may have on our business in the future. Such changes could, among other things, require:
| S-32 |
| • | requests for additional endpoints or studies; |
| • | changes to manufacturing methods; |
| • | recall, replacement, or discontinuance of certain products; and |
| • | additional record keeping. |
Each of these would likely entail substantial time and cost and could have a material adverse effect on our ability to obtain regulatory approval for our product candidates. In addition, delays in receipt of or failure to receive regulatory clearances or approvals for any future products could materially and adversely affect our business, results of operations and financial condition.
Our ability to market AL001, ALZN002 and any future product candidates in the United States, if approved, will be limited to use for the treatment of the indications for which they are approved, and if we want to expand the indications for which we may market AL001, ALZN002 and any future product candidates, we will need to obtain additional FDA approvals, which may not be granted.
We plan to seek full FDA approval in the United States for AL001 and ALZN002 to treat neurodegenerative diseases and psychiatric disorders, including Alzheimer’s. In addition, we have submitted a pre-IND meeting request with the FDA to explore AL001 for the treatment of BD, MDD and PTSD. If AL001 or ALZN002 is approved, the FDA will restrict our ability to market or advertise it for the treatment of indications other than the one for which it is approved, which would limit its use. If we decide to attempt to develop, promote and commercialize new treatment indications and protocols for AL001, ALZN002 and potentially other product candidates in the future, we could not predict when, or if, we would ever receive the approvals required to do so. We would be required to conduct additional studies to support such applications for additional use, which would consume additional resources and may produce results that do not result in FDA approvals. If we do not obtain additional FDA approvals, our ability to expand our business in the United States would be adversely affected, which could materially and adversely affect our business, results of operations and financial condition.
The anticipated development of a REMS for AL001 or ALZN002 could cause delays in the approval process and would add additional layers of regulatory requirements that could impact our ability to commercialize AL001 and ALZN002 in the United States and reduce their market potential.
As a condition of approval of an NDA or a BLA, the FDA may require a REMS to ensure that the benefits of the drug outweigh the potential risks. REMS elements can include medication guides, communication plans for health care professionals, and elements to assure safe use (“ETASU”). ETASU’s can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring, and the use of patient registries. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy. We may be required to adopt a REMS for AL001 or ALZN002 to ensure that the benefits outweigh the risks of abuse, misuse, diversion and other potential safety concerns. Even if the risk of abuse, misuse or diversion are not as high as for some other products, there can be no assurance that the FDA will approve a manageable REMS for AL001 or ALZN002, which could create material and significant limits on our ability to successfully commercialize AL001 and ALZN002 in the U.S. Delays in the REMS approval process could result in delays in the NDA or BLA approval process, respectively. In addition, as part of the REMS, the FDA could require significant restrictions, such as restrictions on the prescription, distribution and patient use of the product, which could significantly impact our ability to effectively commercialize AL001 or ALZN002, and dramatically reduce their market potential thereby adversely impacting our business, financial condition and results of operations. Even if initial REMS are not highly restrictive, if, after launch, AL001, ALZN002 and other drug candidates were to become subject to significant abuse/non-medical use or diversion from licit channels, this could lead to negative regulatory consequences, including a more restrictive REMS, which could materially and adversely affect our business, results of operations and financial condition.
If we are found in violation of “fraud and abuse” laws, we may be subject to criminal and civil penalties and/or be suspended or excluded from participation in government-run health care programs, which may adversely affect our business, financial condition and results of operations.
If we are successful in obtaining marketing approval for our products in the United States and elsewhere, we will be subject to various health care “fraud and abuse” laws, including anti-kickback laws, false claims laws and other laws intended to reduce fraud and abuse in government-run health care programs, which could materially and adversely affect us, particularly upon successful commercialization of our products in the United States. For example, the federal Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf), to knowingly and willfully solicit, receive, offer or pay any remuneration that is intended to induce the referral of business, including the purchase, order or prescription of a particular drug for which payment may be made under a U.S. health care program such as Medicare or Medicaid. Under U.S. federal government regulations, some arrangements, known as safe harbors, are deemed not to violate the Anti-Kickback Statute. Compliance with every element of a safe harbor regulation is required for the arrangement to be protected. However, arrangements that do not comply with a safe harbor are not per se illegal. Instead, they will be analyzed on a case-by-case basis. Although we intend to seek to structure our business arrangements in compliance with all applicable requirements, these laws are broadly written, and it is often difficult to determine precisely how the law will be applied in specific circumstances. Accordingly, it is possible that our practices may be challenged under the Anti-Kickback Statute and similar laws in other jurisdictions.
| S-33 |
Further, false claims laws prohibit anyone from knowingly and willfully presenting or causing to be presented for payment to third-party payers, including government payers, reimbursement claims for drugs or services that are false or fraudulent, claims for items or services that were not provided as claimed, or claims for medically unnecessary items or services. Cases have been brought under false claims laws alleging that off-label promotion of pharmaceutical products or the payment of kickbacks by pharmaceutical providers has resulted in the submission of false claims to governmental health care programs. Under laws such as the Health Insurance Portability and Accountability Act of 1996 in the United States, we are prohibited from knowingly and willfully executing a scheme to defraud any health care benefit program, including private payers, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for health care benefits, items or services. Violations of fraud and abuse laws may be punishable by criminal and/or civil sanctions, including fines and/or exclusion or suspension from government-run health care programs such as Medicare and Medicaid and debarment from contracting with the U.S. and other governments. In addition, in the United States, individuals have the ability to bring actions on behalf of the government and potentially share in the recovery under the federal False Claims Act as well as under state false claims laws.
Many states in the United States have adopted fraud and abuse laws similar to their federal counterparts, including laws similar to the Anti-Kickback Statute, some of which apply to the referral of patients for health care services reimbursed by any source, not just governmental payers. In addition, California and some other states in the United States have passed laws that require pharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America Code on Interactions with Health Care Professionals. In addition, several states impose other marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to the state. There are ambiguities as to what is required to comply with these state requirements and if we fail to comply with an applicable state law requirement, we could be subject to penalties.
We have yet to receive definitive guidance on the application of fraud and abuse laws to our business. Law enforcement authorities are increasingly focused on enforcing these laws, and it is possible that some of our future practices may be challenged under these laws. While we believe we will be able to structure our business arrangements to comply with these laws, it is possible that the government could in the future allege violations of, or convict us of violating, these laws. If we are found in violation of one of these laws, we could be required to pay a penalty and could be suspended or excluded from participation in certain government-run health care programs, and our business, results of operations and financial condition may be materially and adversely affected.
Risks Related to Our Business and Industry
If we fail to attract and keep senior management and key scientific personnel, we may be unable to successfully develop AL001, ALZN002 or any future product candidates, conduct our in-licensing and development efforts or commercialize AL001, ALZN002 or any of our future product candidates.
Our future growth and success depend in part on our continued ability to attract, retain and motivate highly qualified management and scientific personnel. We are highly dependent upon our senior management, particularly Stephan Jackman, our Chief Executive Officer, David J. Katzoff, our Chief Financial Officer, Kenneth S. Cragun, our Senior Vice President of Finance and Henry Nisser, our Executive Vice President and General Counsel. The loss of services of any of these individuals could delay or prevent the successful development of our current or future product pipeline, completion of our planned development efforts or the commercialization of AL001 or ALZN002. It is possible that current or former employees of ours could put forward claims for an alleged right to our patents and demand compensation therefor. If one or more of the key personnel were to leave us and engage in competing operations, our business, results of operations and financial condition could be materially and adversely affected.
We expect to face substantial competition, with other entities possibly discovering, developing or commercializing products before, or more successfully than, we do.
The development, FDA approval and commercialization of new therapy and vaccine products is highly competitive. We will face competition with respect to AL001, ALZN002 and any other product candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. In addition to existing therapeutic treatments for the indications we are targeting with AL001 and ALZN002, we also face potential competition from other drug candidates in development by other companies. Our potential competitors include, without limitation, large health care companies, such as Biogen Inc., Eisai Co., Ltd., Takeda Pharmaceuticals, Bristol Myers Squibb, Pfizer Inc., Merck & Co., Inc., Sanofi S.A., Eli Lilly and Company, Bayer AG, Novartis AG, Johnson and Johnson and Boehringer Ingelheim GmbH. We also know of several smaller early-stage companies that are developing products for use in our segment of the market. Some of the potential competitive compounds referred to above are being developed by large, well-financed and established pharmaceutical and biotechnology companies or have been partnered with such companies, which may give them development, regulatory and marketing advantages over our products.
| S-34 |
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. In addition, our ability to compete may be affected in many cases by insurers or other third-party payers seeking to encourage the use of generic products. If AL001 or ALZN002 achieves marketing approval, we expect that it will be priced at a significant premium over competing generic products.
Some of the companies against which we are competing or against which we may compete in the future have significantly greater financial, physical and human resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller and other early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
If we are unable to compete successfully, we may be unable to grow and sustain our revenue, which could materially and adversely affect our business, results of operations and financial condition.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise prevent our product candidates from being developed or commercialized in a timely manner, which could negatively impact our business.
We rely on the FDA to assist with the development our product candidates. The ability of the FDA to review and approve new drug products can be affected by a variety of factors outside of our control, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the agency have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for our product candidates to be reviewed and/or potentially approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. If the timing of FDA’s review and approval of new products is delayed, the estimated timing of our drug development program may be delayed which would materially increase costs of drug development and harm our operations or business.
Risks Related to Our Intellectual Property
We may be forced to litigate to enforce or defend our intellectual property rights, or the intellectual property rights of our licensors.
We may be forced to litigate to enforce or defend our intellectual property rights against infringement and unauthorized use by competitors. In so doing, we may place our intellectual property at risk of being invalidated, held unenforceable, or narrowed in scope. Further, an adverse result in any litigation or defense proceedings may place pending applications at risk of non-issuance. In addition, if any licensor fails to enforce or defend its intellectual property rights, this may adversely affect our ability to develop and commercialize AL001 or ALZN002 as well as our ability to prevent competitors from making, using, and selling competing products. Any such litigation could be very costly and could distract our management from focusing on operating our business. The existence or outcome of any such litigation could harm our business, results of operations and financial condition.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential and proprietary information could be compromised by disclosure during this type of litigation. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the price of our common stock.
| S-35 |
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We rely on trade secrets to protect our proprietary know-how and technological advances, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights. Failure to obtain or maintain trade secret protection or failure to adequately protect our intellectual property could enable competitors to develop generic products or use our proprietary information to develop other products that compete with our products or cause additional, material adverse effects upon our business, results of operations and financial condition.
The transfer of technology and knowledge to contract manufacturers pursuant to the production of our products also creates a risk of uncontrolled distribution and copying of concepts, methods and processes relating to our products. Such uncontrolled distribution and copying could have a material adverse effect on the value of our products if used for the production of competing drugs or otherwise used commercially without our obtaining financial compensation.
We may become subject to third parties’ claims alleging infringement of patents and proprietary rights or seeking to invalidate our patents or proprietary rights, which would be costly, time-consuming and, if successfully asserted against us, delay or prevent the development and commercialization of AL001 or ALZN002.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical industry, as well as patent challenge proceedings, including interference and administrative law proceedings before the USPTO and the European Patent Office (“EPO”), and oppositions and other comparable proceedings in other jurisdictions. Recently, under U.S. patent reform laws, new procedures including inter partes review and post grant review have been implemented. As stated below, the novel implementation of such laws presents uncertainty regarding the outcome of challenges to our patents in the future.
We cannot assure you that AL001, ALZN002 or any of our future product candidates will not infringe existing or future patents. We may be unaware of patents that have already issued that a third party might assert are infringed by AL001, ALZN002 or one of our future product candidates. Because patent applications can take many years to issue and may be confidential for 18 months or more after filing, there may be applications now pending of which we are unaware of and which may later result in issued patents that we may infringe by commercializing AL001, ALZN002 or any of our future product candidates. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Moreover, we may face claims from non-practicing entities (commonly referred to as patent trolls), which have no relevant product revenue and against whom our own patent portfolio may thus have no deterrent effect.
We may be subject to third-party claims in the future against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing a third party’s patents. If a patent infringement suit were brought against us or our collaborators, we or our collaborators could be forced to stop or delay research, development, manufacturing or sales of AL001 or ALZN002. As a result of patent infringement claims, or in order to avoid potential claims, we or our collaborators may choose to seek, or be required to seek, a license from the third party and would most likely be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which would give our competitors access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or forced to redesign it, or to cease some aspect of our business operations if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms. Even if we are successful in defending such claims, infringement and other intellectual property litigation can be expensive and time-consuming to litigate and divert management’s attention from our core business. Any of these events could harm our business significantly.
In addition to infringement claims against us, if third parties have prepared and filed patent applications in the U.S. that also claim technology to which we have rights, we may have to participate in interference proceedings in the USPTO to determine the priority of invention. Third parties may also attempt to initiate reexamination, post grant review or inter partes review of our patents in the USPTO. We may also become involved in similar opposition proceedings in the EPO or comparable offices in other jurisdictions regarding our intellectual property rights with respect to our products and technology. Any of these claims could have a material adverse effect on our business, results of operations and financial condition.
| S-36 |
If our efforts to protect the proprietary nature of the intellectual property related to AL001, ALZN002 or any of our potential future product candidates are not adequate, we may not be able to compete effectively in our market.
We expect to rely upon a combination of patents, trade secret protection as well as confidentiality and license agreements to protect the intellectual property related to our product and our current product candidates and our development programs.
Composition-of-matter patents on an active pharmaceutical ingredient are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, as such patents provide protection without regard to any particular method of use or manufacture. We cannot be certain that the claims in any patent application that we may submit covering composition-of-matter of AL001, ALZN002 and any potential future product candidates will be considered patentable by the USPTO and courts in the U.S., or by the patent offices and courts in foreign countries. Method-of-use patents protect the use of a product for the specified method. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method.
The strength of patents involves complex legal and scientific questions and can be uncertain. The patent applications that we may in the future own or license may fail to result in issued patents in the United States or in other foreign countries. Even if the patents do successfully issue, third parties may challenge the validity, enforceability or scope thereof, which may result in such patents being narrowed, invalidated or held unenforceable. Furthermore, even if they are unchallenged, any of our future patents and patent applications may not adequately protect our intellectual property or prevent others from designing around our claims. If the breadth or strength of protection provided by the patent applications we may own, license or pursue with respect to AL001, ALZN002 or any future product candidates is threatened, it could threaten our ability to commercialize AL001, ALZN002 or any future product candidates. Further, if we encounter delays in our development efforts, the period of time during which we could market AL001, ALZN002 or any future product candidates under patent protection would be reduced. Since patent applications in the U.S. and most other countries are confidential for a period of time after filing, we cannot be certain that we were the first to file any patent application related to AL001, ALZN002 or any future product candidates.
Even where laws provide protection, costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and the outcome of such litigation would be uncertain. Moreover, any actions we may bring to enforce our intellectual property against our competitors could provoke them to bring counterclaims against us, and some of our competitors have substantially greater intellectual property portfolios than we have.
We will also rely on trade secret protection and confidentiality agreements to protect proprietary know-how that is not patentable, processes for which patents are difficult to enforce and any other elements of our product development processes that involve proprietary know-how, information or technology that is not covered by patents. Although we endeavor to execute confidentiality agreements with all of our employees, consultants, advisors and any third parties who have access to our proprietary know-how, information or technology, we cannot be certain that we have executed such agreements with all parties who may have helped to develop our intellectual property or had access to our proprietary information, nor that our agreements will not be breached. We cannot guarantee that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Further, the laws of some foreign countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States or the European Union. As a result, we may encounter significant problems in protecting and defending our intellectual property not only in the United States and the European Union, but elsewhere as well. If we are unable to prevent material disclosure of the intellectual property related to our technologies to third parties, we will not be able to establish or maintain a competitive advantage in our market, which could materially and adversely affect our business, results of operations and financial condition and any disclosure to or misappropriation by third parties of our confidential proprietary information could enable competitors to quickly duplicate or surpass our technological achievements, thus eroding our competitive position in our market.
Changes in patent law could diminish the value of patents in general, thereby impairing our ability to protect AL001 and ALZN002.
As is the case with other biopharmaceutical companies, our success will be heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity. Therefore, obtaining and enforcing biopharmaceutical patents is costly, time-consuming and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing wide-ranging patent reform legislation. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in other situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents, once obtained. Depending on decisions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in ways that would weaken our ability to obtain patents and to enforce patents that we might obtain in the future. Similarly, changes in EU patent law and elsewhere could negatively affect the value of our patents registered outside of the U.S.
| S-37 |
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with any of these requirements.
The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other provisions during the patent process. There are situations in which noncompliance can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, competitors might be able to enter the market earlier than would otherwise have been the case, which could have a material adverse effect on our business, results of operations and financial condition.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on AL001, ALZN002 and any future product candidates throughout the world is prohibitively expensive. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection, but where enforcement is not as strong as that in the U.S. These products may compete with our products in jurisdictions where we do not have any issued or licensed patents and our patent claims or other intellectual property rights may not be effective or sufficient to prevent them from so competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biopharmaceuticals, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
Risks Relating to Legal Matters
If product liability lawsuits are brought against us, we will incur substantial liabilities and may be required to limit the commercialization of AL001 or ALZN002.
We and our partners face potential product liability exposure related to the testing of AL001 or ALZN002 in clinical trials. We will face exposure to claims by an even greater number of persons if we begin to market and distribute our products commercially in the U.S. and elsewhere, including those relating to misuse of AL001 or ALZN002. Now, and in the future, an individual may bring a liability claim against us alleging that AL001 or ALZN002 caused an injury. While we intend to take what we believe to be appropriate precautions, we may be unable to avoid significant liability if any product liability lawsuit is brought against us. If we cannot successfully defend ourselves against product liability claims, we will incur substantial liabilities. Even if we successfully defend any such action, the costs associated with such defense could prove exorbitant. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for AL001 or ALZN002 (if such product candidate had been approved and gone to market); |
| • | injury to our reputation; |
| • | withdrawal of clinical trial participants; |
| • | costs of related litigation; |
| • | substantial monetary awards to patients and others; |
| • | increased cost of liability insurance; |
| • | loss of revenue; and |
| • | our inability to successfully commercialize our products. |
Further, in the future there may be a need to expand the scope of our insurance coverage, which could result in significantly increased costs or the inability to obtain sufficient insurance coverage. Any of these occurrences could have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Our Affiliates’ Control and Relationships
Insiders currently have substantial influence over us, which could limit your ability to affect the outcome of key transactions, including a change of control.
In the aggregate, beneficial ownership of the shares of our common stock by our directors and executive officers and their respective affiliated parties represents approximately 49.5% of the outstanding shares of our common stock. As a result, these stockholders, if they act together, will be able to influence our management and affairs and all matters requiring stockholder approval, including the election of directors and approval of significant corporate transactions. This concentration of ownership may have the effect of delaying or preventing a change in control of our company and might affect the market price of our common stock.
| S-38 |
Members of the Board of Directors and executive officers of our company and AULT, contain some of the same individuals, which may present potential conflicts of interest.
Our company is controlled by Milton C. (Todd) Ault III, our Founder, Chairman Emeritus and consultant, directly and indirectly through his controlling equity interest in Ault & Company, Inc. the parent of Ault Life Sciences, Inc. (“ALSI”) and Ault Life Sciences Fund, LLC (“ALSF”). Mr. Ault is also the Executive Chairman and single largest stockholder (through his control of Ault Alpha, LP) of AULT, a publicly traded diversified holding company focused primarily on digital mining of Bitcoin and its metaverse platform, oil exploration, crane services, defense/aerospace, industrial, automotive, medical/biopharma, consumer electronics, hotel operations and textiles. The Board of Directors and executive officers of our company and the board of directors and executive officers of AULT contain some of the same individuals, all of whom devote a portion of their business and professional time and efforts to the respective businesses of our company as well as AULT. William B. Horne, the Chairman of the Board of our company, is the Chief Executive Officer and a director of AULT, Henry Nisser, our Executive Vice President, General Counsel and a director of our company, is the President, General Counsel and a director of AULT and Kenneth S. Cragun, our Senior Vice President of Finance is the Chief Financial Officer of AULT.
While we believe that our business and technologies are distinguishable from those of AULT and that we do not compete in the markets in which AULT compete, Mr. Ault and the other named individuals may have potential conflicts of interest with respect to, among other things, potential corporate opportunities, business combinations, joint ventures and/or other business opportunities that may become available to them, our company or AULT. Moreover, while Mr. Ault and the other named individuals have agreed to devote a portion of their business and professional time and efforts to our company, potential conflicts of interest also include the amount of time and effort devoted by each of them to the affairs of AULT. We may be materially adversely affected if Mr. Ault and/or the other named individuals choose to place the interests of AULT before those of our company. Each of Mr. Ault and the other named individuals has agreed that, to the extent such opportunities arise, he will carefully consider a number of factors, including whether such opportunities were presented to him in his capacity as an officer or director of our company, whether such opportunities are within our company’s line of business or consistent with our strategic objectives and whether our company will be able to undertake or benefit from such opportunities. In addition, our Board of Directors has adopted a policy whereby any future transactions between us and any of our affiliates, officers, directors, principal stockholders or any affiliates of the foregoing will be on terms no less favorable to our company than could reasonably be obtained in “arm’s length” transactions with independent third parties, and any such transactions will also be approved by a majority of our disinterested independent directors. The named individuals, other than Mr. Ault, owe fiduciary duties of good faith, care and loyalty to our company under Delaware law. However, the failure of our management to resolve any conflicts of interest in favor of our company could materially adversely affect our business, financial condition and results of operations.
Certain provisions of our certificate of incorporation allow concentration of voting power, which may, among other things, delay or frustrate the removal of incumbent directors or a takeover attempt, even if such events may be beneficial to our stockholders.
Provisions of our certificate of incorporation may delay or frustrate the removal of incumbent directors and may prevent or delay a merger, tender offer or proxy contest involving our company that is not approved by our Board of Directors, even if those events may be perceived to be in the best interests of our stockholders. Further, we may designate and issue separate classes of preferred stock that may entitle their holder(s) to exercise significant control over us. Consequently, anyone to whom or which these shares are or were issued could have sufficient voting power to significantly influence if not control the outcome of all corporate matters submitted to the vote of our common stockholders. Those matters could include the election of directors, changes in the size and composition of our Board, and mergers and other business combinations involving us. In addition, through any such person’s control of our Board and voting power, the affiliate may be able to control certain decisions, including decisions regarding the qualification and appointment of officers, dividend policy, access to capital (including borrowing from third-party lenders and the issuance of additional debt or equity securities), and the acquisition or disposition of assets by us. In addition, the concentration of voting power in the hands of an affiliate could have the effect of delaying or preventing a change in control of our company, even if the change in control could benefit our stockholders and may adversely affect the future market price of our common stock should a trading market therefor develop.
Risks Relating to Ownership of Our Common Stock
If we do not regain compliance with or continue to satisfy the Nasdaq Capital Market continued listing requirements, our common stock could be delisted from the Nasdaq Capital Market.
The listing of our common stock on the Nasdaq Capital Market is contingent on our compliance with the Nasdaq Capital Market’s conditions for continued listing. We are currently not in compliance with Nasdaq listing requirements, specifically the minimum bid price requirement, and must regain compliance on or prior to July 31, 2023. If we are unable to regain such compliance, we will cease to be eligible to trade on Nasdaq and will likely be delisted by Nasdaq.
| S-39 |
If we were to fail to meet a Nasdaq Capital Market listing requirement, we may be subject to delisting by the Nasdaq Capital Market. In the event our common stock is no longer listed for trading on the Nasdaq Capital Market, our trading volume and share price may decrease and we may experience further difficulties in raising capital which could materially affect our operations and financial results. Further, delisting from the Nasdaq Capital Market could also have other negative effects, including potential loss of confidence by partners, lenders, suppliers and employees and could also trigger various defaults under our lending agreements and other outstanding agreements. Finally, delisting could make it harder for us to raise capital and sell securities. You may experience future dilution as a result of future equity offerings. In order to raise additional capital, we may in the future offer additional shares of our common stock or other securities convertible into or exchangeable for our common stock.
We do not know whether an active market will be sustained; as a result, it may be difficult for you to sell your shares of our common stock.
If an active market for our common stock is not sustained, it may be difficult for you to sell your shares of common stock at an attractive price or at all. We cannot predict the prices at which our common stock will trade. It is possible that in one or more future periods our results of operations and progression of our product pipeline may not meet the expectations of public market analysts and investors and, as a result of these and other factors, the price of our common stock may fall.
The market price of our common stock is volatile, which could result in substantial losses for investors.
Our common stock is listed on the Nasdaq Capital Market. Since our initial public offering last year, our trading price has fluctuated widely, depending on many factors that may have little to do with our operations or business prospects. During the year ended April 30, 2023, our stock closed at prices between $0.425 per share and $1.19 per share, as reported on Nasdaq.com.
Stock markets, in general, have experienced, and continue to experience, significant price and volume volatility, and the market price of our common stock may continue to be subject to similar market fluctuations unrelated to our operating performance or prospects. This increased volatility, coupled with depressed economic conditions, could continue to have a depressive effect on the market price of our common stock. The following factors, many of which are beyond our control, may influence our stock price:
| • | announcements of the failure to obtain regulatory approvals or receipt of a “complete response letter” from the FDA; |
| • | announcements of restricted label indications or patient populations, or changes or delays in regulatory review processes; |
| • | announcements of therapeutic innovations or new products by us or our competitors; |
| • | adverse actions taken by regulatory agencies with respect to our clinical trials, manufacturing supply chain or sales and marketing activities; |
| • | changes or developments in laws or regulations applicable to our product candidates; |
| • | any failure of our testing and clinical trials; |
| • | product liability claims, other litigation or public concern about the safety of our product candidates or future products; |
| • | any adverse changes to our relationship with licensors, manufacturers or suppliers; |
| • | the loss of any of our key scientific or management personnel; |
| • | any major changes to our Board of Directors or management; |
| • | the failure to obtain new commercial partners; |
| • | announcements concerning our competitors or the pharmaceutical industry in general; |
| • | the failure to achieve expected product sales and profitability; |
| • | the failure to obtain reimbursements for our product candidates as part of any healthcare insurance plan, or reductions in such reimbursements; |
| • | actual or anticipated fluctuations in our cash position or operating results; |
| • | manufacturing, supply or distribution shortages related to our current or future product candidates for our development programs and commercialization; |
| S-40 |
| • | changes in financial estimates or recommendations by securities analysts; |
| • | the termination of any of our existing license agreements; |
| • | announcements relating to future licensing or development agreements; |
| • | potential acquisitions; |
| • | the trading volume of shares on The Nasdaq Capital Market; |
| • | sales of our shares by us, our executive officers or directors or our shareholders; |
| • | fluctuations in the U.S. equity markets; |
| • | changes in accounting principles; |
| • | market conditions in the healthcare sector; and |
| • | general economic conditions in the United States and elsewhere. |
In recent years, each of the stock market in general, and the market for pharmaceutical and biotechnology companies in particular, has experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. Following periods of such volatility in the market price of a company’s securities, securities class action litigation has often been brought against that company. Because of the potential volatility of our stock price, we may become the target of securities litigation in the future. Securities litigation could result in substantial costs and divert management’s attention and resources from our business.
If there are substantial sales of shares of our common stock, the price of our common stock could decline.
The price of our common stock could decline if there are substantial sales of our common stock, particularly sales by our directors, executive officers and significant stockholders, or if there is a large number of shares of our common stock available for sale and the market perceives that sales will occur. As of July 24, 2023, we had 96,940,124 shares of our common stock outstanding. Shares held by directors, executive officers and other affiliates will be subject to volume limitations under Rule 144 under the Securities Act and various vesting agreements. We have registered shares of common stock that we have issued and may issue under our employee equity incentive plans, which shares may be sold freely in the public market upon issuance. Sales of our common stock by current stockholders may make it more difficult for us to sell equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate, and make it more difficult for other stockholders to sell shares of our common stock.
The market price of the shares of our common stock could decline as a result of the sale of a substantial number of our shares of common stock in the public market or the perception in the market that the holders of a large number of shares intend to sell their shares. We are unable to predict the effect that sales may have on the prevailing market price of our common stock.
The concentration of our stock ownership will limit your ability to influence corporate matters, including the ability to influence the outcome of director elections and other matters requiring stockholder approval.
Our executive officers, directors and the holders of more than 5% of our outstanding common stock, in the aggregate, beneficially own a significant percentage of our common stock. As a result, these stockholders, acting together, will have significant influence over all matters that require approval by our stockholders, including the election of directors and approval of significant corporate transactions. Corporate actions might be taken even if other stockholders oppose them. This concentration of ownership might also have the effect of delaying or preventing a change of control of our company that other stockholders may view as beneficial.
Our bylaws provide that the Court of Chancery of the State of Delaware and the federal district courts of the United States are the exclusive forums for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our bylaws provides that the Court of Chancery of the State of Delaware will be the exclusive forum for any derivative action or proceeding brought on our behalf; any action asserting a breach of fiduciary duty; any action asserting a claim against us arising under the Delaware General Corporation Law, our certificate of incorporation or our bylaws; any action to interpret, apply, enforce or determine the validity of our certificate of incorporation or our bylaws; and any action asserting a claim against us that is governed by the internal affairs doctrine. This provision would not apply to suits brought to enforce a duty or liability created by the Exchange Act or any other claim for which the U.S. federal courts have exclusive jurisdiction.
| S-41 |
Our bylaws further provide that the federal district courts of the United States will be the exclusive forum for resolving any complaint asserting a cause of action arising under the Securities Act. The enforceability of similar exclusive federal forum provisions in other companies’ organizational documents has been challenged in legal proceedings, and while the Delaware Supreme Court has ruled that this type of exclusive federal forum provision is facially valid under Delaware law, there is uncertainty as to whether other courts would enforce such provisions and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder.
These exclusive forum provisions may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. Alternatively, if a court were to find either exclusive forum provision in our bylaws to be inapplicable or unenforceable in an action, we may incur further significant additional costs associated with resolving such action in other jurisdictions, all of which could have a material adverse effect on our business, financial condition, and results of operations.
General Risk Factors
We must effectively manage the growth of our operations, or our company will suffer.
Our initiation of operations has resulted in significantly higher operating expenses. Expansion of our operations, to include the development of AL001 and ALZN002, may also cause a significant demand on our management, finances and other resources. Our ability to manage the anticipated future growth, should it occur, will depend upon a significant expansion of our accounting and other internal management systems and the implementation and subsequent improvement of a variety of systems, procedures and controls. In addition, we intend to expand our scientific advisory board. There can be no assurance that significant problems in these areas will not occur. Any failure to expand these areas and implement and improve AL001 or ALZN002 or our procedures and controls in an efficient manner at a pace consistent with our business could have a material adverse effect on our business, financial condition and results of operations. There can be no assurance that our attempts to expand our marketing, sales, manufacturing and customer support efforts will be successful or will result in additional sales or profitability in any future period.
We may not be successful in our efforts to expand our pipeline of product candidates.
One element of our strategy is to expand our pipeline of pharmaceuticals based on our technology and advance these product candidates through clinical development for the treatment of a variety of indications. Although our research and development efforts to date have resulted in a number of development programs based on our technology, we may not ultimately be able to develop product candidates that are safe and effective. Even if we are successful in continuing to expand our pipeline, the potential product candidates that we identify may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to receive marketing approval and achieve market acceptance. In addition, if we attempt to apply our technology to develop product candidates for indications outside of Alzheimer’s, we will need to evaluate the preclinical data and determine if additional data are needed to support the new indications. If we do not successfully develop and commercialize product candidates based upon our technological approach, we will not be able to obtain product revenue in future periods, which would make it unlikely that we would ever achieve profitability.
We may experience product recalls or inventory losses caused by unforeseen events, cold chain interruption and testing difficulties.
AL001 and ALZN002, individually, will be manufactured and distributed, if ever, using technically complex processes requiring specialized facilities, highly specific raw materials and other production constraints. The complexity of these processes, as well as the strict company and government standards for the manufacture of our products, will subject us to production risks. While product batches released for use in clinical trials or for commercialization undergo sample testing, some defects may only be identified following product release. In addition, process deviations or unanticipated effects of approved process changes may result in these intermediate products not complying with stability requirements or specifications. Most of our products must be stored and transported at temperatures within a certain range, which is known as “strict cold chain” storage and transportation. If these environmental conditions deviate from the norm, our products’ remaining shelf lives could be impaired or their quality could become adversely affected, making them no longer suitable for use. The occurrence or suspected occurrence of production and distribution difficulties can lead to lost inventories, and in some cases product recalls, with consequential reputational damage and the risk of product liability. The investigation and remediation of any identified problems can cause production delays, substantial expense, lost sales and delays of new product launches, any of which could have a material adverse effect on our business, results of operations and financial condition.
| S-42 |
Because we do not intend to pay dividends on our common stock, you must rely on stock appreciation for any return on your investment.
We presently intend to retain any future earnings and do not expect to pay any dividends in the foreseeable future. As a result, you must rely on stock appreciation and a liquid trading market for any return on your investment. If an active and liquid trading market does not develop, you may be unable to sell your shares of common stock at or above the initial public offering price or at the time you would like to sell.
We have identified a material weakness in our internal control over financial reporting. If our remediation of this material weakness is not effective, or if we experience additional material weaknesses in the future or otherwise fail to maintain an effective system of internal controls in the future, we may not be able to accurately or timely report our financial condition or results of operations, which may adversely affect investor confidence in us and, as a result, the value of our common stock.
We have limited accounting personnel to adequately execute our accounting processes and other supervisory resources with which to address our internal control over financial reporting. In connection with the audit of our financial statements for the year ended April 30, 2023, we identified material weaknesses in our internal control over financial reporting. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our financial statements will not be prevented or detected on a timely basis. The material weaknesses related to a lack of sufficient number of qualified personnel within our accounting function to adequately segregate duties, to perform sufficient reviews and approval of manual journal entries posted to the general ledger and to consistently execute review procedures over general ledger account reconciliations, financial statement preparation and accounting for non-routine transactions and, we have not designed and implemented effective Information Technology General Controls (“ITGC”) related to access controls to payment and financial accounting systems.
We are implementing measures designed to improve our internal control over financial reporting to remediate this material weakness, including the following:
| • | We are formalizing our internal control documentation and strengthening supervisory reviews by our management; |
| • | We are in the process of adding additional accounting personnel and segregating duties amongst accounting personnel; and |
| • | We are in the process of strengthening ITGC access controls related to our payment and financial accounting systems. |
We cannot assure you that the measures we have taken to date, and are continuing to implement, will be sufficient to remediate the material weakness we have identified or avoid potential future material weaknesses. If the steps we take do not correct the material weakness in a timely manner, we will be unable to conclude that we maintain effective internal control over financial reporting. Accordingly, there could continue to be a reasonable possibility that a material misstatement of our financial statements would not be prevented or detected on a timely basis.
As a public company, we are required to maintain internal control over financial reporting and to report any material weaknesses in such internal controls. We must perform system and process evaluation and testing of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act. The Sarbanes-Oxley Act also requires that our management report on internal control over financial reporting be attested to by our independent registered public accounting firm, to the extent we are no longer an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012 (JOBS Act). We do not expect our independent registered public accounting firm to attest to our management report on internal control over financial reporting for so long as we are an emerging growth company.
We are in the process of enhancing our internal control over financial reporting required to comply with this obligation, which process will be time consuming, costly and complicated. If we identify any additional material weaknesses in our internal control over financial reporting, if we are unable to comply with the requirements of Section 404 in a timely manner, if we are unable to assert that our internal control over financial reporting is effective, or when required in the future, if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, investors may lose confidence in the accuracy and completeness of our financial reports and the market price of our common stock could be adversely affected, and we could become subject to investigations by the Nasdaq Stock Market, the SEC, or other regulatory authorities, which could require additional financial and management resources.
We may have trouble hiring additional qualified personnel.
As we expand our development and commercial activities, we will need to hire additional personnel and could experience difficulties attracting and retaining qualified employees. Competition for qualified personnel in the biopharmaceutical field is intense due to the limited number of individuals who possess the skills and experience required by that industry. We may not be able to attract and retain quality personnel on favorable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that such personnel have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output. Any of these difficulties could have a material adverse effect on our business, results of operations and financial condition.
| S-43 |
Failure of our information technology systems could significantly disrupt the operation of our business.
Our ability to execute our business plan and to comply with regulatory requirements with respect to data control and data integrity depends, in part, on the continued and uninterrupted performance of our information technology systems, or IT systems. These systems are vulnerable to damage from a variety of sources, including telecommunications or network failures, malicious human acts and natural disasters. Moreover, despite network security and back-up measures, some of our servers are potentially vulnerable to physical or electronic break-ins, computer viruses and similar disruptive problems. Despite the precautionary measures we have taken to prevent unanticipated problems that could affect our IT systems, there are no assurances that electronic break-ins, computer viruses and similar disruptive problems, and/or sustained or repeated system failures or problems arising during the upgrade of any of our IT systems that interrupt our ability to generate and maintain data will not occur. The occurrence of any of the foregoing with respect to our IT systems could have a material adverse effect on our business, results of operations or financial condition.
We are subject to various claims and legal actions arising in the ordinary course of our business.
We are subject to various claims and legal actions arising in the ordinary course of our business. Any such litigation could be very costly and could distract our management from focusing on operating our business. The existence of any such litigation could harm our business, results of operations and financial condition. Results of actual and potential litigation are inherently uncertain. An unfavorable result in a legal proceeding could adversely affect our reputation, financial condition and operating results.
We will be subject to the U.S. Foreign Corrupt Practices Act and other anti-corruption laws, as well as export control laws, customs laws, sanctions laws and other laws governing our anticipated operations. If we fail to comply with these laws, we could be subject to civil or criminal penalties, other remedial measures, and legal expenses, which could adversely affect our business, results of operations and financial condition.
Our operations, if initiated, will be subject to certain anti-corruption laws, including the U.S. Foreign Corrupt Practices Act (“FCPA”), and other anti-corruption laws that apply in countries where we do business. The FCPA and other anti-corruption laws generally prohibit us and our employees and intermediaries from bribing, being bribed or making other prohibited payments to government officials or other persons to obtain or retain business or gain some other business advantage. We and any future commercial partners may operate in a number of jurisdictions that pose a high risk of potential FCPA violations and we may participate in collaborations and relationships with third parties whose actions could potentially subject us to liability under the FCPA or local anti-corruption laws. In addition, we cannot predict the nature, scope or effect of future regulatory requirements to which our international operations might be subject or the manner in which existing laws might be administered or interpreted.
We also anticipate becoming subject to other laws and regulations governing our international operations, including regulations administered in the U.S. and in the EU, including applicable export control regulations, economic sanctions on countries and persons, customs requirements and currency exchange regulations (collectively, “Trade Control Laws”).
There can be no assurance that we will be completely effective in ensuring our compliance with all applicable anticorruption laws, including the FCPA or other legal requirements, such as Trade Control Laws. Any investigation of potential violations of the FCPA, other anti-corruption laws or Trade Control Laws by the United States, the European Union or other authorities could have an adverse impact on our reputation, our business, results of operations and financial condition. Furthermore, should we be found not to be in compliance with the FCPA, other anti-corruption laws or Trade Control Laws, we may be subject to criminal and civil penalties, disgorgement and other sanctions and remedial measures, as well as the accompanying legal expenses, any of which could have a material adverse effect on our reputation and liquidity, as well as on our business, results of operations and financial condition.
Certain provisions of our certificate of incorporation, bylaws and Delaware law make it more difficult for a third party to acquire us and make a takeover more difficult to complete, even if such a transaction were in the stockholders’ interest.
Our certificate of incorporation, bylaws and certain provisions of Delaware law could have the effect of making it more difficult or more expensive for a third party to acquire, or discouraging a third party from attempting to acquire, control of our company, even when these attempts may be in the best interests of our stockholders. For example, we are governed by Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. A “business combination” includes mergers, asset sales or other transactions resulting in a financial benefit to the stockholder. An “interested stockholder” is a person who, together with affiliates and associates, owns, or within three years did own, 15% or more of the corporation’s outstanding voting stock. These provisions may have the effect of delaying, deferring or preventing a change in control of our company.
| S-44 |
Failure to build our finance infrastructure and improve our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, the regulations of The Nasdaq Capital Market, the rules and regulations of the SEC, expanded disclosure requirements, accelerated reporting requirements and more complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud. We must perform system and process evaluation and testing of our internal controls over financial reporting to allow management to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act.
We anticipate that the process of building our accounting and financial functions and infrastructure will require significant additional professional fees, internal costs and management efforts. We expect that we will need to implement a new internal system to combine and streamline the management of our financial, accounting, human resources and other functions. However, such a system would likely require us to complete many processes and procedures for the effective use of the system or to run our business using the system, which may result in substantial costs. Any disruptions or difficulties in implementing or using such a system could adversely affect our controls and harm our business. Moreover, such disruption or difficulties could result in unanticipated costs and diversion of management attention. In addition, we may discover weaknesses in our system of internal financial and accounting controls and procedures that could result in a material misstatement of our financial statements. Our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
If we are not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner, or if we are unable to maintain proper and effective internal controls, we may not be able to produce timely and accurate financial statements. If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed, investors could lose confidence in our reported financial information and we could be subject to sanctions or investigations by The Nasdaq Capital Market, the SEC or other regulatory authorities.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our stock, the price of our common stock could decline.
The trading market for our common stock will rely in part on the research and reports that industry or financial analysts publish about us or our business. We do not currently have and may never obtain research coverage by industry or financial analysts. If no or few analysts commence coverage of us, the trading price of our common stock could decrease. Even if we do obtain analyst coverage, if one or more of the analysts covering our business downgrade their evaluations of our stock, the price of our common stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our common stock, which in turn could cause our stock price to decline.
We are an “emerging growth company,” and the reduced disclosure requirements applicable to emerging growth companies may make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. For so long as we remain an emerging growth company, we are permitted and plan to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not emerging growth companies. These exemptions include not being required to comply with the auditor attestation requirements of SOX Section 404, not being required to comply with any requirement that may be adopted by the Public Company Accounting Oversight Board regarding mandatory audit firm rotation or a supplement to the auditor’s report providing additional information about the audit and the financial statements, reduced disclosure obligations regarding executive compensation, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. As a result, the information we provide stockholders will be different than the information that is available with respect to other public companies. In this Annual Report, we have not included all of the executive compensation-related information that would be required if we were not an emerging growth company. We cannot predict whether investors will find our common stock less attractive if we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile.
| S-45 |
We will incur increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives and corporate governance practices.
As a public company, and particularly after we are no longer an emerging growth company (or, to a lesser extent, a smaller reporting company), we will incur significant legal, accounting, and other expenses that we did not incur as a private company. Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of The Nasdaq Capital Market, and other applicable securities rules and regulations impose various requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and corporate governance practices. We expect that we will need to hire additional accounting, finance, and other personnel in connection with our becoming, and our efforts to comply with the requirements of being, a public company, and our management and other personnel will need to devote a substantial amount of time towards maintaining compliance with these requirements. These requirements will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect that the rules and regulations applicable to us as a public company may make it more difficult and more expensive for us to obtain director and officer liability insurance, which could make it more difficult for us to attract and retain qualified members of our Board of Directors. We are currently evaluating these rules and regulations and cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Our charter provides for limitations of director liability and indemnification of directors and officers and employees.
Our certificate of incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
| • | breach of their duty of loyalty to us or our stockholders; |
| • | act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
| • | unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or |
| • | transaction from which the directors derived an improper personal benefit. |
These limitations of liability do not apply to liabilities arising under the federal or state securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our bylaws provide that we will indemnify our directors, officers and employees to the fullest extent permitted by law. Our bylaws also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding. We believe that these provisions are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability in our certificate of incorporation and bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
We could be subject to securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biopharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
| S-46 |
USE OF PROCEEDS
We may issue and sell shares of our common stock having aggregate sales proceeds of up to $9,831,947 from time to time. Because there is no minimum offering amount required as a condition to close this offering, the actual total public offering amount, commissions, expenses, and proceeds to us, if any, are not determinable at this time but will be reported in our periodic reports.
We intend to use the use the net proceeds, if any, from this offering for general corporate purposes, which may include working capital, capital expenditures, research and development expenditures, regulatory affairs expenditures, clinical trial expenditures, acquisitions of new technologies and investments, the financing of possible acquisitions or business expansions, and the repayment, refinancing, redemption or repurchase of future indebtedness or capital stock. We do not have agreements or commitments for any specific acquisitions at this time.
The timing and amount of our actual expenditures will be based on many factors, including cash flows from operations and the anticipated growth of our business. As of the date of this prospectus supplement, we cannot specify with certainty all of the particular uses for the net proceeds to us from this offering. As a result, our management will have broad discretion regarding the timing and application of the net proceeds from this offering. Pending these uses, we intend to invest the net proceeds from this offering in short-term, investment-grade, interest-bearing securities.
Any portion of the $9,831,947 included in this prospectus supplement not previously sold or included in an active placement notice pursuant to the sales agreement, may be later made available for sale in other offerings pursuant to the accompanying base prospectus, and if no shares have been sold under the sales agreement, the full $9,831,947 of shares of common stock may be later made available for sale in other offerings pursuant to the accompanying base prospectus.
| S-47 |
PLAN OF DISTRIBUTION
We have entered into a sales agreement with ACM, under which we may issue and sell over a period of time, and from time to time, shares of our common stock having an aggregate gross sales price of up to $9,831,947 from time to time through ACM acting as a sales agent or directly to ACM acting as principal. Sales of our common stock, if any, under this prospectus may be made in sales deemed to be “at the market offerings” as defined in Rule 415 under the Securities Act, including sales made directly on or through the Nasdaq Capital Market (“Nasdaq”), the trading market for our common stock, or any other trading market in the Unites States for our common stock, sales made to or through a market maker other than on an exchange, directly to the Sales Agent as principal for its account in negotiated transactions at market prices prevailing at the time of sale or at prices related to such prevailing market prices, in privately negotiated transactions, in block trades, or through a combination of any such methods of sale. To the extent required by Regulation M, ACM acting as our sales agent will not engage in any transactions that stabilize our common stock while the offering is ongoing under this prospectus supplement. The sales agreement has been filed as an exhibit to our Current Report on Form 8-K filed with the United States Securities and Exchange Commission (the “SEC”) on September 8, 2023, which is incorporated by reference in this prospectus supplement.
Each time we wish to issue and sell common stock, we will notify ACM of the number of shares to be issued, the dates on which such sales are anticipated to be made, any minimum price below which sales may not be made and other sales parameters as we deem appropriate, subject to certain limitations set forth by the SEC. Once we have so instructed ACM, unless ACM declines to accept the terms of the notice, ACM has agreed, subject to the terms and conditions of the sales agreement, to use its commercially reasonable efforts consistent with its normal trading and sales practices to sell such shares up to the amount specified on such terms. We may instruct ACM not to sell shares of common stock if the sales cannot be effected at or above the price designated by us in any such instruction. ACM may also sell our common stock in negotiated transactions with our prior approval. We or ACM may suspend the offering of shares of common stock being made through ACM under the sales agreement upon proper notice to the other party.
We will pay ACM commissions for its services in acting as agent in the sale of our common stock. ACM will be entitled to compensation at a commission rate equal to 3.5% of the aggregate gross sales price of the shares sold. The remaining sales proceeds, after deducting any expenses payable by us and any transaction fees imposed by any governmental, regulatory or self-regulatory organization in connection with the sales, will equal our net proceeds for the sale of such shares. Because there is no minimum offering amount in this offering, the actual total public offering amount, commissions and proceeds to us, if any, are not determinable at this time. We have also agreed to reimburse ACM for certain specified expenses, including the fees and disbursements of its legal counsel in an amount not to exceed $10,000 and, thereafter, the reasonable fees and expenses of ACM’s legal counsel up to $5,000 incurred in connection with quarterly and annual bring-downs required thereunder, as provided in the sales agreement.
Settlement for sales of common stock will occur, unless the parties agree otherwise, on the second business day following the date on which any sales are made, or on some other date that is agreed upon by us and ACM in connection with a particular transaction, in return for payment of the net proceeds to us. There is no arrangement for funds to be received in an escrow, trust or similar arrangement.
In connection with the sale of the common stock on our behalf, ACM will be deemed to be an “underwriter” within the meaning of the Securities Act and the compensation of ACM will be deemed to be underwriting commissions or discounts. We have agreed to provide indemnification and contribution to ACM against certain civil liabilities, including liabilities under the Securities Act. The offering of our common stock pursuant to the sales agreement will terminate upon the earlier of (1) the sale of all shares of our common stock subject to the sales agreement having an aggregate offering price of $10,000,000 (unless the parties agree to extend the sales agreement) or (2) termination of the sales agreement as permitted therein. We may terminate the sales agreement at any time upon five days’ prior notice and ACM may terminate the sales agreement at any time upon ten days’ prior notice.
Our common stock is traded on the Nasdaq Capital Market under the symbol “ALZN.” The transfer agent of our common stock is Computershare Trust Company, N.A., 8742 Lucent Blvd., Suite 225, Highlands Ranch, CO 80129.
Any portion of the $9,831,947 included in this prospectus supplement not previously sold or included in an active placement notice pursuant to the sales agreement, may be later made available for sale in other offerings pursuant to the accompanying base prospectus, and if no shares have been sold under the sales agreement, the full $9,831,947 of shares of common stock may be later made available for sale in other offerings pursuant to the accompanying base prospectus.
ACM and/or its affiliates may also in the future provide various investment banking and other financial services for us for which services they may in the future receive customary fees. We may agree with ACM in the future agree to add one or more additional sales agents to the offering, in which case we will file a further prospectus supplement providing the name of such additional sales agents and any other required information.
This summary of the material provisions of the sales agreement does not purport to be a complete statement of its terms and conditions.
| S-48 |
DESCRIPTION OF OUR SECURITIES
The summary does not purport to be complete and is qualified in its entirety by reference to our certificate of incorporation and bylaws, and to the provisions of the General Corporation Law of the State of Delaware, as amended.
We are authorized to issue 300,000,000 shares of common stock, par value $0.0001 per share. As of the date of this prospectus, there were 96,940,124 shares of our common stock issued and outstanding. The outstanding shares of our common stock are validly issued, fully paid and nonassessable.
We are authorized to issue up to 10,000,000 shares of preferred stock, par value $0.0001 per share. Of these shares of preferred stock, 1,360,000 are designated as Series A Convertible Preferred Stock. As of the date of this prospectus, there were no shares of Series A Convertible Preferred Stock issued or outstanding.
Common Stock
Holders of our shares of common stock are entitled to one vote for each share on all matters submitted to a shareholder vote. Holders of our common stock do not have cumulative voting rights. Therefore, holders of a majority of the shares of our common stock voting for the election of directors can elect all of the directors. Holders of our common stock representing a majority of the voting power of our capital stock issued, outstanding and entitled to vote, represented in person or by proxy, are necessary to constitute a quorum at any meeting of shareholders. A vote by the holders of a majority of our outstanding shares is required to effectuate certain fundamental corporate changes such as liquidation, merger or an amendment to our certificate of incorporation.
Holders of our common stock are entitled to share in all dividends that our board of directors, in its discretion, declares from legally available funds. In the event of a liquidation, dissolution or winding up, each outstanding share entitles its holder to participate pro rata in all assets that remain after payment of liabilities and after providing for each class of stock, if any, having preference over our common stock. Our common stock has no preemptive, subscription or conversion rights and there are no redemption provisions applicable to our common stock.
Preferred Stock
The shares of preferred stock may be issued in series, and shall have such voting powers, full or limited, or no voting powers, and such designations, preferences and relative participating, optional or other special rights, and qualifications, limitations or restrictions thereof, as shall be stated and expressed in the resolution or resolutions providing for the issuance of such stock adopted from time to time by the board of directors. The board of directors is expressly vested with the authority to determine and fix in the resolution or resolutions providing for the issuances of preferred stock the voting powers, designations, preferences and rights, and the qualifications, limitations or restrictions thereof, of each such series to the full extent now or hereafter permitted by the laws of the State of Delaware.
The authorized shares of preferred stock will be available for issuance without further action by our stockholders unless such action is required by applicable law or the rules of any stock exchange or automated quotation system on which our securities may be listed or traded. The Nasdaq Stock Market currently requires stockholder approval as a prerequisite to listing shares in several circumstances, including, in certain circumstances, where the issuance of shares could result in an increase in the number of shares of common stock outstanding, or in the amount of voting securities outstanding, of at least 20%.
Shares Offered in this Prospectus Supplement
We are offering up to $13 million in shares of our common stock pursuant to this prospectus supplement.
Transfer Agent and Registrar
The Transfer Agent and Registrar for our common stock is Computershare Trust Company, N.A., 8742 Lucent Blvd., Suite 225, Highlands Ranch, CO 80129.
| S-49 |
LEGAL MATTERS
Olshan Frome Wolosky LLP, New York, New York, as our counsel, will pass upon the validity of the common stock offered by this prospectus supplement and accompanying prospectus. Clyde Snow & Sessions, P.C., Salt Lake City, Utah, is acting as counsel to the sales agent in connection with certain legal matters relating to this offering.
EXPERTS
The financial statements of Alzamend Neuro, Inc. as of April 30, 2023 and 2022 and for each of the two years in the period ended April 30, 2023 incorporated by reference in this Prospectus and Registration Statement from our Annual Report on Form 10-K for the years ended April 30, 2023 and 2022, have been audited by Baker Tilly US, LLP, an independent registered public accounting firm, as stated in their report thereon (which report expresses an unqualified opinion and includes an explanatory paragraph relating to the Company’s ability to continue as a going concern), incorporated herein by reference, and have been incorporated in this Prospectus and Registration Statement in reliance upon such report and upon the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
This prospectus supplement and the accompanying prospectus are part of the registration statement on Form S-3 we filed with the SEC under the Securities Act, and do not contain all the information set forth in the registration statement. Whenever a reference is made in this prospectus supplement or the accompanying prospectus to any of our contracts, agreements or other documents, the reference may not be complete, and you should refer to the exhibits that are a part of the registration statement or the exhibits to the reports or other documents incorporated by reference into this prospectus supplement and the accompanying prospectus for a copy of such contract, agreement or other document. You may inspect a copy of the registration statement, including the exhibits and schedules, without charge, at the SEC's public reference room mentioned below, or obtain a copy from the SEC upon payment of the fees prescribed by the SEC.
We file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read, without charge, and copy the documents we file at the SEC’s public reference rooms in Washington, D.C. at 100 F Street, NE, Room 1580, Washington, DC 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for further information on the public reference rooms. Our SEC filings are also available to the public at no cost from the SEC’s website at http://www.sec.gov.
INCORPORATION OF DOCUMENTS BY REFERENCE
We have filed a registration statement on Form S-3 with the Commission under the Securities Act. This prospectus is part of the registration statement but the registration statement includes and incorporates by reference additional information and exhibits. The Commission permits us to “incorporate by reference” the information contained in documents we file with the Commission, which means that we can disclose important information to you by referring you to those documents rather than by including them in this prospectus. Information that is incorporated by reference is considered to be part of this prospectus and you should read it with the same care that you read this prospectus. Information that we file later with the Commission will automatically update and supersede the information that is either contained, or incorporated by reference, in this prospectus, and will be considered to be a part of this prospectus from the date those documents are filed. We have filed with the Commission, and incorporate by reference in this prospectus:
| • | Our Annual Report on Form 10-K for the year ended April 30, 2023, as filed with the SEC on July 27, 2023; |
| • | Our Current Reports on Form 8-K, as filed with the SEC on August 7, 2023 and September 8, 2023; and |
| • | Our Definitive Proxy Statement on Schedule 14A, as filed with the SEC on August 24, 2023. |
We also incorporate by reference all additional documents that we file with the Securities and Exchange Commission under the terms of Sections 13(a), 13(c), 14 or 15(d) of the Exchange Act that are made after the initial filing date of the registration statement of which this prospectus is a part until the offering of the particular securities covered by a prospectus supplement or term sheet has been completed. We are not, however, incorporating, in each case, any documents or information that we are deemed to furnish and not file in accordance with Commission rules.
We will provide you, without charge upon written or oral request, a copy of any and all of the information that has been incorporated by reference in this prospectus and that has not been delivered with this prospectus. Requests should be directed to Alzamend Neuro, Inc., 3480 Peachtree Road NE, Second Floor, Suite 103, Atlanta, GA 30326; Tel: (844) 722-6333; Attention: Mr. Stephan Jackman, Chief Executive Officer.
| S-50 |
Filed pursuant to Rule 424(b)(1)
Registration No. 333-273610
$25,000,000
Common Stock
Preferred Stock
Warrants
Rights
Units
We may offer and sell, from time to time in one or more offerings, any combination of common stock, preferred stock, warrants, rights or units having an aggregate initial offering price not exceeding $25,000,000. The preferred stock, warrants, rights and units may be convertible, exercisable or exchangeable for common stock or preferred stock or other securities of ours.
Each time we sell a particular class or series of securities, we will provide specific terms of the securities offered in a supplement to this prospectus. The prospectus supplement may also add, update or change information in this prospectus. You should read this prospectus and any prospectus supplement, as well as the documents incorporated by reference or deemed to be incorporated by reference into this prospectus, carefully before you invest in any securities.
This prospectus may not be used to offer or sell our securities unless accompanied by a prospectus supplement relating to the offered securities.
Our common stock is presently listed on the Nasdaq Capital Market under the symbol “ALZN.” On July 31, 2023, the last reported sale price of our common stock was $0.453.
These securities may be sold directly by us, through dealers or agents designated from time to time, to or through underwriters or dealers or through a combination of these methods on a continuous or delayed basis. See “Plan of Distribution” in this prospectus. We may also describe the plan of distribution for any particular offering of our securities in a prospectus supplement. If any agents, underwriters or dealers are involved in the sale of any securities in respect of which this prospectus is being delivered, we will disclose their names and the nature of our arrangements with them in a prospectus supplement. The net proceeds we expect to receive from any such sale will also be included in a prospectus supplement.
An investment in our common stock involves a high degree of risk. You should review carefully the risks and uncertainties described under the heading “Risk Factors” contained on page 11 of this prospectus and in our Annual Report on Form 10-K for the year ended April 30, 2023, as well as our subsequently filed periodic and current reports that we file with the Securities and Exchange Commission and which are incorporated by reference into the registration statement of which this prospectus is a part. We may also include additional risk factors in a prospectus supplement under the heading “Risk Factors.” You should read this prospectus and the applicable prospectus supplement carefully before you make your investment decision.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
This prospectus is dated August 10, 2023
TABLE OF CONTENTS
|
Page
| ||
| About this Prospectus | 1 | |
| Disclosure Regarding Forward-Looking Statements | 1 | |
| Prospectus Summary | 2 | |
| Risk Factors | 11 | |
| Use of Proceeds | 11 | |
| Plan of Distribution | 11 | |
| Description of Securities We May Offer | 13 | |
| Description of Capital Stock | 14 | |
| Description of Warrants | 15 | |
| Description of Rights | 17 | |
| Description of Units | 17 | |
| Legal Matters | 18 | |
| Experts | 18 | |
| Where you can find more Information | 18 | |
| Incorporation of Documents by Reference | 19 |
| i |
ABOUT THIS PROSPECTUS
This prospectus is part of a shelf registration statement that we filed with the Securities and Exchange Commission (the “Commission”) using a “shelf” registration process. Under this shelf registration process, we may sell any combination of the securities described in this prospectus in one or more offerings from time to time having an aggregate initial offering price of $25,000,000. This prospectus provides you with a general description of the securities we may offer. Each time we offer securities, we will provide you with a prospectus supplement that describes the specific amounts, prices and terms of the securities we offer. The prospectus supplement also may add, update or change information contained in this prospectus. You should read carefully both this prospectus and any prospectus supplement together with additional information described below under the caption “Where You Can Find More Information.”
This prospectus does not contain all the information provided in the registration statement we filed with the Commission. You should read both this prospectus, including the section titled “Risk Factors,” and the accompanying prospectus supplement, together with the additional information described under the heading “Where You Can Find More Information.”
This prospectus may be supplemented from time to time to add, to update or change information in this prospectus. Any statement contained in this prospectus will be deemed to be modified or superseded for purposes of this prospectus to the extent that a statement contained in such prospectus supplement modifies or supersedes such statement. Any statement so modified will be deemed to constitute a part of this prospectus only as so modified, and any statement so superseded will be deemed not to constitute a part of this prospectus. You should rely only on the information contained or incorporated by reference in this prospectus, any applicable prospectus supplement or any related free writing prospectus. We have not authorized any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. No dealer, salesperson or other person is authorized to give any information or to represent anything not contained in this prospectus, any applicable prospectus supplement or any related free writing prospectus. This prospectus is not an offer to sell securities, and it is not soliciting an offer to buy securities, in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this prospectus or any prospectus supplement, as well as information we have filed with the Commission that is incorporated by reference, is accurate as of the date on the front of those documents only, regardless of the time of delivery of this prospectus or any applicable prospectus supplement, or any sale of a security. Our business, financial condition, results of operations and prospects may have changed since those dates.
No person is authorized in connection with this prospectus to give any information or to make any representations about us, the securities offered hereby or any matter discussed in this prospectus, other than the information and representations contained in this prospectus. If any other information or representation is given or made, such information or representation may not be relied upon as having been authorized by us.
This prospectus contains summaries of certain provisions contained in some of the documents described herein, but reference is made to the actual documents for complete information. All of the summaries are qualified in their entirety by the actual documents. Copies of some of the documents referred to herein have been filed, will be filed or will be incorporated by reference as exhibits to the registration statement of which this prospectus is a part, and you may obtain copies of those documents as described below under “Where You Can Find More Information.”
For investors outside the United States: Neither we nor any underwriter has done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. You are required to inform yourselves about and to observe any restrictions relating to this offering and the distribution of this prospectus.
Unless otherwise stated or the context requires otherwise, references to “Alzamend,” the “Company,” “we,” “us” or “our” are to Alzamend Neuro, Inc., a Delaware corporation.
DISCLOSURE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the documents incorporated by reference in it contain forward-looking statements regarding future events and our future results that are subject to the safe harbors created under the Securities Act of 1933 and the Securities Exchange Act of 1934. All statements other than statements of historical facts are statements that could be deemed forward-looking statements. These statements are based on our expectations, beliefs, forecasts, intentions and future strategies and are signified by the words “expects,” “anticipates,” “intends,” “believes” or similar language. In addition, any statements that refer to projections of our future financial performance, our anticipated growth, trends in our business and other characterizations of future events or circumstances are forward-looking statements. These forward-looking statements are only predictions and are subject to risks, uncertainties and assumptions that are difficult to predict, including those identified above, under “Risk Factors” and elsewhere in this prospectus. Therefore, actual results may differ materially and adversely from those expressed in any forward-looking statements. All forward-looking statements included in this prospectus are based on information available to us on the date of this prospectus and speak only as of the date hereof.
| 1 |
We disclaim any current intention to update our “forward-looking statements,” and the estimates and assumptions within them, at any time or for any reason. In particular, the following factors, among others, could cause actual results to differ materially from those described in the “forward-looking statements”:
| • | our need for substantial additional funding to finance our operations and complete development to seek FDA approval for AL001 and ALZN002 before commercialization; |
| • | our ability to effectively execute our business strategy; |
| • | our ability to manage our expansion, growth and operating expenses; |
| • | our ability to evaluate and measure our business, prospects and performance metrics; |
| • | our ability to compete and succeed in a highly competitive and evolving industry; |
| • | our ability to respond and adapt to changes in technology and customer behavior; |
| • | our ability to protect our intellectual property and to develop, maintain and enhance a strong brand; |
| • | our significant losses since inception and anticipation that we will continue to incur significant losses for the foreseeable future; |
| • | our reliance on licenses from a third party regarding our rights and development of AL001 and AL002; |
| • | our development of AL001 and AL002 never leading to a marketable product; |
| • | our product candidates not qualifying for expedited development, or if they do, not actually leading to a faster development or regulatory review or approval process; |
| • | our approach to targeting beta-amyloid plaque via AL002 being based on a novel therapeutic approach; and |
| • | the risk factors included in our most recent filings with the SEC, including, but not limited to, our Forms 10-K and 10-Q, which are incorporated by reference herein. |
PROSPECTUS SUMMARY
This summary highlights selected information contained in other parts of this prospectus. Because it is a summary, it does not contain all of the information that you should consider in making your investment decision. Before investing in our securities, you should read the entire prospectus carefully, including the information set forth under the heading “Risk Factors.”
We are a clinical-stage biopharmaceutical company focused on developing novel products for the treatment of Alzheimer’s disease (“Alzheimer’s”), bipolar disorder (“BD”), major depressive disorder (“MDD”) and post-traumatic stress disorder (“PTSD”). With our two product candidates, we aim to bring treatments or potential cures to market as quickly as possible. Far too many individuals, patients and caregivers suffer from the burden created by these devastating, and often fatal, diseases. Our primary target, Alzheimer’s, was among the most-feared diseases (second only to cancer) among Americans, according to a 2011 survey by the Harvard School of Public Health. Alzheimer’s is also the seventh leading cause of death in the United States (“U.S.”) according to a 2021 report from the Alzheimer’s Association, a nonprofit that funds research. Existing Alzheimer’s treatments only temporarily relieve symptoms and while one treatment has been shown to slow the progression of the disease, no treatments have been shown to halt the progression of the disease, which currently affects roughly 6.7 million Americans and that number is expected to grow to 13 million individuals by 2050. Alzheimer’s also impacts more than 11 million Americans who provide an estimated 16 billion hours of unpaid care per year, valued at $272 billion, according to data provided by the Alzheimer’s Association. In 2022, the estimated healthcare costs for treating individuals with Alzheimer’s in the U.S. will be $321 billion, including $206 billion in Medicare and Medicaid payments. These costs could rise to as high as $1 trillion per year by 2050 if no permanent treatment or cure for Alzheimer’s is found, the Alzheimer’s Association reported.
Our pipeline consists of two novel therapeutic drug candidates:
| · | AL001 - A patented ionic cocrystal technology delivering a therapeutic combination of lithium, salicylate and proline through three royalty-bearing exclusive worldwide licenses from the University of South Florida Research Foundation, Inc., as licensor (the “Licensor”); and |
| 2 |
| · | ALZN002 - A patented method using a mutant peptide sensitized cell as a cell-based therapeutic vaccine that seeks to restore the ability of a patient’s immunological system to combat Alzheimer’s through a royalty-bearing exclusive worldwide license from the Licensor. |
Our most advanced product candidate (lead product) licensed and in clinical development in humans is AL001, an ionic cocrystal of lithium for the treatment of Alzheimer’s, BD, MDD and PTSD. Based on our preclinical data involving mice models, AL001 treatment prevented cognitive deficits, depression and irritability and is superior in improving associative learning and memory and irritability compared with lithium carbonate treatments, supporting the potential of this lithium formulation for the treatment of Alzheimer’s, BD, MDD and PTSD in humans. Lithium has been marketed for more than 35 years and human toxicology regarding lithium use has been well characterized, potentially mitigating the regulatory burden for safety data.
The results of randomized, placebo-controlled, clinical trials of lithium in the treatment of patients with Alzheimer’s dementia and subjects with mild cognitive impairment have been widely published. Clinical studies have indicated that lithium administered at doses lower than those used for affective disorders can favorably impact Alzheimer’s outcomes. A study by O.V. Forlenza, et al., entitled “Disease-Modifying Properties of Long-Term Lithium Treatment for Amnestic Mild Cognitive Impairment: Randomized Controlled Trial,” appearing in the British Journal of Psychiatry (2011) reported that lithium was superior to a placebo, evidencing a slower decline of cognitive function as measured by the Alzheimer’s Disease Assessment Scale cognitive subscale. Given the absence of adequate widely adapted treatments that can slow, halt or even reverse the decline of this highly prevalent disease, the potential efficacy of lithium in the long-term management of Alzheimer’s may positively impact public health. There is an unmet medical need for safe and effective Alzheimer’s treatments, particularly for treatments with neuroprotective properties.
There is increasing evidence to suggest that depressive illness, particularly in the elderly, is associated with neuronal cell loss. These findings suggest that lithium may exert some of its long-term beneficial effects in the treatment of affective disorders via underappreciated neuroprotective effects. Molecular biology and animal studies have also suggested that lithium may offer protection against Alzheimer’s. Given the absence of other adequate treatments, the potential efficacy of lithium in the long-term treatment of neurodegenerative disorders may be warranted.
Our Business Strategy
We intend to develop and commercialize therapeutics that are better than existing treatments and have the potential to significantly improve the lives of individuals afflicted by Alzheimer’s, BD, MDD and PTSD. To achieve these goals, we are pursuing the following key business strategies:
| • | Advance clinical development of AL001 for Alzheimer’s, BD, MDD and PTSD treatment. We completed our Phase I clinical trial in March 2022 and initiated a Phase IIA MAD clinical trial in May 2022. We completed the clinical portion of the Phase IIA Multiple Ascending Dose (“MAD”) clinical trial in March 2023 and reported topline data in June 2023. We intend to initiate two Phase II clinical trials to investigate the safety and efficacy of AL001 for patients with mild to moderate Alzheimer’s. Additionally, we intend to investigate the potential of AL001 for patients suffering from BD, MDD and PTSD by submitting investigational new drug (“IND”) applications to the U.S. Food and Drug Administration (“FDA”) for these indications by the end of 2023. If we achieve successful Phase III clinical trials in humans, we intend to seek approval to commercialize AL001 via a New Drug Application (“NDA”); |
| • | Advance clinical development of ALZN002 for Alzheimer’s treatment. We submitted an IND application to the FDA in September 2022, and received a “study may proceed” letter in October 2022. In April 2023, we initiated a Phase I/IIA clinical trial for ALZN002 to treat mild to moderate dementia of the Alzheimer’s type. If we achieve successful Phase III clinical trials in humans, we intend to seek approval to commercialize ALZN002 via an NDA; |
| • | Expand our pipeline of pharmaceuticals to include additional indications for AL001 and delivery methods. Another element of our business strategy is to expand our pipeline of pharmaceuticals based on our technology and advance these product candidates through clinical development for the treatment of a variety of indications. In addition to treating Alzheimer’s, AL001 has the potential to treat a wide range of neurodegenerative diseases and psychiatric disorders. We plan to pursue the treatment of BD, MDD, and PTSD with AL001, and in May 2022, we submitted a pre-Investigational New Drug (“pre-IND”) meeting request to the FDA for these indications and received a written response from the FDA in July 2022. Based on the written response from the FDA and the receipt of topline data from the Phase IIA MAD clinical trial, we plan to submit separate INDs for BD, MDD, and PTSD by the end of 2023, which, after receipt from the FDA of a “study may proceed” letter for such indication, would allow us to initiate a Phase II study. We also plan to explore different formulations (liquid, immediate release and sprinkle capsules) to deliver AL001; |
| 3 |
| • | Focus on translational and functional endpoints to efficiently develop product candidates. We believe AL001 is positioned for a Section 505(b)(2) regulatory pathway for new drug approvals. We also believe AL001 and ALZN002 are positioned for breakthrough therapy designations because of their positive effects on a pharmacodynamic biomarker (beta-amyloids) and potential for a clinically meaningful effect on Alzheimer’s, making them eligible to receive assistance from the FDA throughout the development process that may shorten the development timelines. However, we have neither received breakthrough therapy designation nor have we qualified for expedited development, and no assurance can be given that we will. Even if we qualify for breakthrough therapy designation or expedited development, it may not actually lead to faster development or expedited regulatory review and approval or necessarily increase the likelihood that we will receive FDA approval; and |
| • | Optimize the value of AL001 and ALZN002 in major markets. We intend to commercialize AL001 and ALZN002 by seeking FDA marketing approval for both product candidates and partnering with biopharmaceutical companies seeking to strategically fortify pipelines and, in turn, receiving funding for the costly later-stage clinical development. We do not anticipate selling products directly into the marketplace, though we may do so depending on market conditions. Our focus is expected to concentrate on entering into strategical transactions with established distributors and producers, which will provide distribution and marketing capabilities for the sale of our products into the marketplace. |
Our Development Pipeline
The following chart provides an overview of the current development stages of our therapeutic product candidates.
Our product candidates will require extensive clinical evaluation, regulatory review and approval, significant marketing efforts and substantial investment before it or any successors are likely to provide us with any revenue. As a result, if we do not successfully develop, achieve regulatory approval for and commercialize our product candidates, our long-term business plans will not be met, and we will be unable to generate the revenue we have forecast for the foreseeable future, if any. We do not anticipate that we will generate our maximum revenue for several years, or that we will achieve profitability for any of our therapeutic drug candidates until at least a few years after generating material revenue, if at all. If we are unable to generate revenue or raise substantial additional capital, we will not be able to pursue any expansion of our business or acquire additional intellectual property, we will not become profitable with our therapeutic drug candidates, and we will be unable to continue our operations at the currently planned pace, if at all.
AL001 Drug Candidate
Our lead product candidate that we have licensed and have begun clinical development in humans is an ionic cocrystal of lithium for the treatment of Alzheimer’s, BD, MDD and PTSD. Lithium salts have a long history of human consumption beginning in the 1800s. In psychiatry, they have been used to treat mania and as a prophylactic for depression since the mid-20th century. Today, lithium salts are used as a mood stabilizer for the treatment of BD. Although the FDA has approved no medications as safe and effective treatments for suicidality, lithium has proven to be the only drug that consistently reduces suicidality in patients with neuropsychiatric disorders. Despite these effective medicinal uses, current FDA-approved lithium pharmaceutics (lithium carbonate and lithium citrate) are limited by a narrow therapeutic window that requires regular blood monitoring of plasma lithium levels and blood chemistry by a clinician to mitigate adverse events. Because conventional lithium salts (carbonate and citrate) are eliminated relatively quickly, multiple administrations throughout the day are required to safely reach therapeutic plasma concentrations. Existing lithium drugs, such as lithium chloride and lithium carbonate, suffer from chronic toxicity, poor physicochemical properties and poor brain bioavailability. Because lithium is so effective at reducing manic episodes in patients with BD, it is still used clinically despite its narrow therapeutic index. This has led researchers to begin to look for alternatives to lithium with similar bioactivities.
Scientists from the University of South Florida have developed a new lithium cocrystal composition and method of preparation that, under certain clinical and/or testing conditions, have been shown to allow for lower dosages to achieve therapeutic brain levels of lithium for psychiatric disorders, which could lead to a broadening of lithium’s therapeutic index. Our studies and/or testing have indicated that the compound offers improved physiochemical properties compared to existing forms of lithium, giving it the potential to be developed as an anti-suicidal drug and for use against mood disorders.
| 4 |
Recent evidence suggests that lithium may be efficacious for both the treatment and prevention of Alzheimer’s. Unlike traditional medications which only address a single therapeutic target, lithium appears to be neuroprotective through several modes of action. For example, recent studies have indicated that it exerts neuroprotective effects, in part, by increasing a brain-derived neurotrophic factor leading to restoration of learning and memory. Another neuroprotective mechanism of lithium indicated by recent studies is the attenuation of the production of inflammatory cytokines like IL-6 and nitric oxide in activated microglia. Results from recent clinical studies suggest that lithium treatment may reduce dementia development while preserving cognitive function and reducing biomarkers associated with Alzheimer’s.
The novel ionic cocrystal of lithium (AL001), which was designed, synthesized and characterized by a team of inventors from the University of South Florida has been shown to exhibit improved nonclinical pharmacokinetics compared to currently FDA-approved lithium products and is also bioactive in many in vitro models of Alzheimer’s. AL001 may constitute a means of treating Alzheimer’s, BD, MDD and PTSD.
We believe that our ability to re-engineer lithium solid dosage forms in order to optimize performance and has the potential to address a wide range of clinical applications ranging from neurodegenerative disorders, not merely Alzheimer’s, but also amyotrophic lateral sclerosis (known as ALS and Lou Gehrig’s disease), Huntington’s disease, multiple sclerosis, Parkinson’s disease and traumatic brain injury, to more psychiatric conditions such as BD, MDD, mania, PTSD and suicidality. This novel approach is intended to achieve the desired therapeutic outcome of enhanced penetration through the blood-brain barrier and sustained brain lithium concentrations while systemic exposures (and toxicities) are mitigated for other organ systems. The optimal modified-release lithium dosing approach for AL001 should avoid acutely toxic peak concentrations in blood, as well as in the brain, and should maintain such blood concentrations for a predictable, clinically relevant time, with overall low systemic exposures that mitigate the potential for adverse events. We anticipate that the lithium delivery system will be adaptable to a dosing regimen that maintains therapeutic brain lithium concentrations consistently for the longest possible time while allowing only modest exposures and providing adequate recovery periods between doses for other organ systems.
Clinical Trials
Phase I Study
On September 13, 2021, we initiated a randomized, balanced, Phase I, single-dose, open-label, two-treatment, two-period, two- sequence, crossover, relative bioavailability clinical trial to investigate lithium pharmacokinetics and safety of AL001 formulation compared to a marketed immediate release lithium carbonate formulation in healthy subjects. The primary objective of this clinical trial was to assess the relative bioavailability of the AL001 lithium formulation relative to a marketed lithium carbonate formulation in healthy subjects for the purpose of determining potential clinically safe and effective AL001 dosing in future studies. Additionally, we wanted to characterize safety and tolerability of the tested formulations under the conditions of this clinical trial. This was a first-in-human clinical trial of the AL001 formulation; this trial was designed to assess the relative bioavailability of the AL001 lithium formulation compared to a marketed lithium carbonate formulation in at least 24 completed healthy subjects (30 subjects were to be enrolled) for the purpose of determining potential clinically safe and effective AL001 dosing in future clinical trials. The AL001 lithium content was nearly half of the reference lithium carbonate capsule dosage as it was expected that treatment of frail Alzheimer’s patients will require half the lithium dose used for treatment of BD. Lithium carbonate 300 mg (Reference product) was given as a single dose in this clinical trial; this is often used as a starting dose for treatment of BD when given three times daily. The shape of the AL001 lithium plasma concentration versus time curve was unknown prior to this study. Also unknown were the AL001 rate and extent of lithium absorption. The Phase I study was completed in March 2022 with the following results:
| · | AL001 was shown to be safe and well-tolerated in healthy adult subjects; |
| · | No serious adverse events and no deaths were reported during the trial; |
| · | The safety profiles of both AL001 and the marketed lithium carbonate capsule were benign; |
| · | No clinically significant abnormal findings in electrocardiograms were noted during the trial; |
| · | AL001 salicylate plasma concentrations were observed to be well tolerated and consistently within safe limits; and |
| · | Dose-adjusted relative bioavailability analyses of the rate and extent of lithium absorption in plasma indicated that AL001, at a lithium carbonate equivalent dose of 150 mg, is bioequivalent to a marketed 300 mg lithium carbonate capsule and the shapes of the lithium plasma concentration versus time curves are similar. |
| 5 |
Phase IIA Study
On May 5, 2022, we initiated a multiple-dose, steady-state, double-blind, ascending dose safety, tolerability, pharmacokinetic clinical trial (www.clinicaltrials.gov, identifier: NCT05363293) of AL001 in patients with mild to moderate Alzheimer’s and healthy subjects with the following objectives:
| · | Primary: To evaluate the safety and tolerability of AL001 under multiple-dose, steady-state conditions in Alzheimer’s patients and healthy subjects; |
| · | Secondary: To characterize the maximum tolerated dose (MTD) of AL001 in patients with mild to moderate Alzheimer’s and healthy subjects; and |
| · | Exploratory: Determination of qualitative and quantitative evaluations of AD patient and healthy subjects desirable characteristics for future Phase II and III clinical studies in order to: |
| o | Facilitate recruitment into subsequent AL001 clinical trials; and |
| o | Facilitate trial-adherence to completion of study requirements including treatment adherence. |
We completed the Phase IIA clinical trial in March 2023 and announced positive topline data in June 2023. We announced that we successfully identified a maximum tolerated dose (“MTD”) for development of AL001 from a multiple-ascending dose study as assessed by an independent safety review committee. This dose, providing lithium at a lithium carbonate equivalent dose of 240 mg 3-times daily (“TID”), is designed to be unlikely to require lithium therapeutic drug monitoring (“TDM”). Also, this MTD is risk mitigated for the purpose of treating fragile populations, such as Alzheimer’s patients.
Lithium is a commonly prescribed drug for manic episodes in BP type 1 as well as maintenance therapy of BP in patients with a history of manic episodes. Lithium is also prescribed off-label for MDD, BP and treatment of PTSD, among other disorders. Lithium was the first mood stabilizer approved by the FDA and is still a first-line treatment option (considered the “gold standard”) but is underutilized perhaps because of the need for TDM. Lithium was the first drug that required TDM by regulatory authorities in product labelling because the effective and safe range of therapeutic drug blood concentrations is narrow and well defined for treatment of BP when using lithium salts. Excursions above this range can be toxic, and below can impair effectiveness.
Planned Future Studies
Based on the results from our Phase IIA MAD study, we plan to initiate two safety and efficacy clinical trials in subjects with mild to moderate dementia of the Alzheimer’s type. Additionally, we intend to investigate the potential of AL001 for patients suffering from BD, MDD and PTSD by submitting IND applications to the FDA for these indications by the end of 2023. After FDA permission to proceed on the INDs, we intend to initiate clinical trials at this MTD to determine relative increased lithium levels in the brain compared to a marketed lithium salt for BD, MDD and PTSD, based on published mouse studies that predict that lithium can be given at lower doses for equivalent therapeutic benefit when treating with AL001. For example, the goal is to replace a 300 mg TID lithium carbonate dose for treatment of BD with a 240 mg TID AL001 lithium equivalent, which represents a daily decrease of 20% of lithium given to a patient.
ALZN002 Drug Candidate
The other product candidate that we have licensed to clinically develop in humans is ALZN002, a patented method using a mutant peptide sensitized cell as a cell-based therapeutic vaccine which seeks to restore the ability of the patient’s immunological system to combat Alzheimer’s. The proposed mechanism of action is through the pulsed-Dendritic Cell (“DC”) activation of T-cells that stimulates the immune system, resulting in the clearance of brain amyloid. Preclinical studies conducted from April 2005 to July 2010 demonstrated that the infusion of transgenic (or genetically modified) mice with ALZN002-pulsed DCs is associated with lower amyloid burden and improved neuro-behavioral performance. This is likely to be mediated by an anti-inflammatory effect in addition to the immunogenicity of this therapy.
ALZN002 is based on the theory that Alzheimer’s symptoms may be caused in large part by plaque deposits that can cluster in the brain composed of protein fragments called beta-amyloids that build up between nerve cells. One hypothesis is that a special type of immune cell, natural beta-amyloid antibodies, may play a role in preventing plaque build-up in people without Alzheimer’s. As people age, their immune systems may degrade, and some people may be unable to produce natural beta-amyloid antibodies, the absence of which leads to the plaque build-up causing Alzheimer’s.
ALZN002 is intended to elicit an immune response to produce anti-amyloid antibodies, which can then neutralize circulated beta-amyloids and prevent additional plaque build-up. The mutant antigen within ALZN002 was selected specifically for its high Human Leukocyte Antigens (“HLA”) binding affinity, thereby avoiding the need for an adjuvant, which may cause an adverse (Th1) immune response.
| 6 |
ALZN002 is an autologous modified DC treatment. More precisely, it is a patient-specific therapy where the patient undergoes leukapheresis, a nonsurgical treatment used to reduce the quantity of white blood cells in the bloodstream, to isolate peripheral blood monocytes that are subsequently matured into DCs using cytokine therapy (IL4+ GM-CSF) cocktail. The DCs are incubated with a modified amyloid beta (Aβ) peptide to sensitize them, and then administered to the same patient.
Significant evidence has accumulated recently suggesting that immunotherapy is a highly promising modality of treatment in Alzheimer’s. Most current immune-based active investigations are focused on passive immunization by pre-prepared Aβ antibody administration. Active immunization may offer additional or more lasting effects on the clearance of amyloid and a safer approach due to its reliance on autologous immune mechanisms. Further, preliminary evidence suggests a recurrence of the amyloid accumulation after clearance with the immunoglobulins. A prior attempt at engaging the immune system to treat Alzheimer’s was conducted using the immunization with pre-aggregated synthetic Aβ (AN-1792) combined with the immunogenic adjuvant QS-21. The Phase IIA study with AN-1792 was terminated by the FDA due to severe meningoencephalitis in approximately 6% of vaccinated subjects. We believe that this may have been caused by using a QS-21 adjuvant in the vaccine formulation.
Clinical Trials
Pre-Clinical
On July 23, 2021, we announced that Alzamend received positive toxicology results for ALZN002 in a good laboratory practices (“GLP”) toxicology study using a transgenic mouse model of Alzheimer’s. The study was conducted by Charles River Laboratories. ALZN002 is a patented method using a mutant-peptide sensitized cell as a cell-based therapeutic vaccine that seeks to restore the ability of a patient’s immunological system to combat Alzheimer’s.
A five-dose GLP study with ALZN002-sensitized cells was completed using a transgenic (or genetically modified) mouse model of Alzheimer’s to investigate the tolerability of ALZN002. Single injections were administered on days 1, 30, 50, 70, and 90. The mice were evaluated for potential toxicity and reversibility of any findings at 75 and 90 days after dosing.
Histopathology results demonstrate that there was no indication of T-cell infiltration or meningoencephalitis suggesting that ALZN002 therapy is safe and tolerable as there were no adverse findings over a 90-day period and 90 days after the last dose. There were no treatment-related mortalities or reports of adverse effects on clinical observations, body weight parameters, organ weight parameters, clinical pathology parameters, gross pathology observations, or histopathologic observations during the main study or the recovery phase.
Modified cell therapies, especially DCs, may provide a safer and more patient-specific active immunization. Ex-vivo modification of DCs as a modality of treatment has been previously used in oncological therapeutics. It has been shown to be relatively safe and capable of engaging the immune system to attack the target tissues with success. Its use in Alzheimer’s therapeutics is relatively recent.
Phase I/II Study
We submitted a pre-IND meeting request for ALZN002 and supporting briefing documents to the Center for Biological Evaluation and Research of the FDA on July 30, 2021. We received a written response relating to the pre-IND from the FDA providing a path for Alzamend’s planned clinical development of ALZN002 on September 30, 2021. The FDA agreed to allow Alzamend to submit an IND to conduct a combined Phase I/II study.
On September 28, 2022, we submitted an IND application to the FDA for ALZN002 and received a “study may proceed” letter on October 31, 2022. The product candidate is an immunotherapy vaccine designed to treat mild to moderate dementia of the Alzheimer’s type. ALZN002 is a proprietary “active” immunotherapy product, which means it is produced by each patient’s immune system. It consists of autologous DCs that are activated white blood cells taken from each individual patient so that they can be engineered outside of the body to attack Alzheimer’s-related amyloid-beta proteins. These DCs are pulsed with a novel amyloid-beta peptide (E22W) designed to bolster the ability of the patient’s immune system to combat Alzheimer’s; the goal being to foster tolerance to treatment for safety purposes while stimulating the immune system to reduce the brain’s beta-amyloid protein burden, resulting in reduced Alzheimer’s signs and symptoms. Compared to passive immunization treatment approaches that use foreign blood products (such as monoclonal antibodies), active immunization with ALZN002 is anticipated to offer a more robust and long-lasting effect on the clearance of amyloid. This could provide a safer approach due to its reliance on autologous immune components, using each individual patient’s own white blood cells rather than foreign cells and/or blood products.
On April 3, 2023, we announced the initiation of a phase I/IIA clinical trial for ALZN002 to treat mild to moderate dementia of the Alzheimer’s type. The purpose of this trial is to assess the safety, tolerability, and efficacy of multiple ascending doses of ALZN002 compared with that of a placebo in 20-30 subjects with mild to moderate morbidity. The primary goal of this clinical trial is to determine an appropriate dose of ALZN002 for treatment of patients with Alzheimer’s in a larger Phase IIB efficacy and safety clinical trial, which Alzamend expects to initiate within three months of receiving data from the initial trial.
| 7 |
The continuation of our current development plans with respect to completing our IND applications and conducting the series of human clinical trials for each of our therapeutics requires us to raise additional capital to fund our operations.
Intellectual Property and Licensing Agreements
On July 2, 2018, we entered into two Standard Exclusive License Agreements with Sublicensing Terms for AL001 with the Licensor and its affiliate, the University of South Florida (the “AL001 Licenses”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide licenses limited to the field of Alzheimer’s, under U.S. Patent Nos. (i) 9,840,521, entitled “Organic Anion Lithium Ionic Cocrystal Compounds and Compositions”, filed September 24, 2015 and granted December 12, 2017, and (ii) 9,603,869, entitled “Lithium Co-Crystals for Treatment of Neuropsychiatric Disorders”, filed May 21, 2016 and granted March 28, 2017. On February 1, 2019, we entered into the First Amendment to the AL001 Licenses, on March 30, 2021, we entered into the Second Amendment to the AL001 Licenses and on June 8, 2023, we entered into the Third Amendment to the AL001 Licenses (collectively, the “AL001 License Agreements”).
The AL001 License Agreements require that we pay combined royalty payments of 4.5% on net sales of products developed from the licensed technology for AL001. We have already paid an initial license fee of $200,000 for AL001. As an additional licensing fee for the license of the AL001 technologies, the Licensor received 2,227,923 shares of our common stock. Minimum royalties for AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the AL001 License Agreements.
On May 1, 2016, we entered into a Standard Exclusive License Agreement with Sublicensing Terms for ALZN002 with the Licensor (the “ALZN002 License”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide license limited to the field of Alzheimer’s Immunotherapy and Diagnostics, under U.S. Patent No. 8,188,046, entitled “Amyloid Beta Peptides and Methods of Use”, filed April 7, 2009 and granted May 29, 2012. On August 18, 2017, we entered into the First Amendment to the ALZN002 License, on May 7, 2018, we entered into the Second Amendment to the ALZN002 License, on January 31, 2019, we entered into the Third Amendment to the ALZN002 License, on January 24, 2020, we entered into the Fourth Amendment to the ALZN002 License, on March 30, 2021, we entered into the Fifth Amendment to the ALZN002 License and on April 17, 2023, we entered into the Sixth Amendment to the ALZN002 License (collectively, the “ALZN002 License Agreement”).
The ALZN002 License Agreement requires us to pay royalty payments of 4% on net sales of products developed from the licensed technology for ALZN002. We have already paid an initial license fee of $200,000 for ALZN002. As an additional licensing fee for the license of ALZN002, the Licensor received 3,601,809 shares of our common stock. Minimum royalties for ALZN002 are $20,000 on the first anniversary of the first commercial sale, $40,000 on the second anniversary first commercial sale and $50,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the ALZN002 License Agreement.
On November 19, 2019, we entered into two Standard Exclusive License Agreements with Sublicensing Terms for two additional indications of AL001 with the Licensor (the “November AL001 License”), pursuant to which the Licensor granted us a royalty bearing exclusive worldwide licenses limited to the fields of (i) neurodegenerative diseases excluding Alzheimer’s and (ii) psychiatric diseases and disorders. On March 30, 2021, we entered into the First Amendments to the November AL001 License and on April 17, 2023, we entered into the Second Amendments to the November AL001 License (collectively, the “November AL001 License Agreements”).
The November AL001 License Agreements require us to pay royalty payments of 3% on net sales of products developed from the licensed technology for AL001 in those fields. We paid an initial license fee of $20,000 for the additional indications. Minimum royalties for November AL001 License Agreements are $40,000 on the first anniversary of the first commercial sale, $80,000 on the second anniversary first commercial sale and $100,000 on the third anniversary of the first commercial sale and every year thereafter, for the life of the November AL001 License Agreements.
These license agreements have an indefinite term that continue until the later of the date no licensed patent under the applicable agreement remains a pending application or enforceable patent, the end date of any period of market exclusivity granted by a governmental regulatory body, or the date on which the licensee’s obligations to pay royalties expire under the applicable license agreement. Under our various license agreements, if we fail to meet a milestone by its specified date, Licensor may terminate the license agreement. The Licensor was also granted a preemptive right to acquire such shares or other equity securities that may be issued from time to time by us while the Licensor remains the owner of any equity securities of our company.
| 8 |
Additionally, we are required to pay milestone payments on the due dates to the Licensor for the license of the AL001 technologies and for the ALZN002 technology, as follows:
Original AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Completed September 2019 | Pre-IND meeting | |
| $ | 65,000 | * | Completed June 2021 | ND application filing | |
| $ | 190,000 | * | Completed December 2021 | Upon first dosing of patient in a clinical trial | |
| $ | 500,000 | * | Completed March 2022 | Upon Completion of first clinical trial | |
| $ | 1,250,000 | 24 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 8 years from the effective date of the agreement | Upon FDA NDA approval | ||
| * | Milestone met and completed |
| 9 |
ALZN002 License:
| Payment | Due Date | Event | |||
| $ | 50,000 | * | Upon IND application filing | Upon IND application filing | |
| $ | 50,000 | September 2023 | Upon first dosing of patient in first Phase I clinical trial | ||
| $ | 500,000 | 24 months from completion of first Phase I clinical trial | Upon completion of first Phase II clinical trial | ||
| $ | 1,000,000 | 12 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 10,000,000 | 7 years from the effective date of the agreement | Upon FDA Biologics License Application (“BLA”) approval | ||
| * | Milestone met and completed |
Additional AL001 Licenses:
| Payment | Due Date | Event | |||
| $ | 2,000,000 | 36 months from completion of the first Phase II clinical trial | Upon first patient treated in a Phase III clinical trial | ||
| $ | 16,000,000 | August 1, 2029 | First commercial sale | ||
Corporate Information
Our principal executive offices are located at 3480 Peachtree Road NE, Second Floor, Suite 103, Atlanta, GA 30326, and our telephone number is (844) 722-6333. Our corporate website address is www.alzamend.com. The information contained on or accessible through our website is not a part of this prospectus.
| 10 |
RISK FACTORS
Investing in our securities involves a high degree of risk. Before deciding whether to invest in our securities, you should carefully consider the risk factors we describe in any prospectus supplement and in any related free writing prospectus for a specific offering of securities, as well as those incorporated by reference into this prospectus and any prospectus supplement. You should also carefully consider other information contained and incorporated by reference in this prospectus and any applicable prospectus supplement, including our financial statements and the related notes thereto incorporated by reference in this prospectus. The risks and uncertainties described in the applicable prospectus supplement and our other filings with the Commission incorporated by reference herein are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently consider immaterial may also adversely affect us. If any of the described risks occur, our business, financial condition or results of operations could be materially harmed. In such case, the value of our securities could decline and you may lose all or part of your investment.
USE OF PROCEEDS
Except as otherwise provided in the applicable prospectus supplement, we intend to use the net proceeds from the sale of the securities offered by this prospectus for general corporate purposes, which may include working capital, capital expenditures, research and development expenditures, regulatory affairs expenditures, clinical trial expenditures, acquisitions of new technologies and investments, the financing of possible acquisitions or business expansions, and the repayment, refinancing, redemption or repurchase of future indebtedness or capital stock.
The intended application of proceeds from the sale of any particular offering of securities using this prospectus will be described in the accompanying prospectus supplement relating to such offering. The precise amount and timing of the application of these proceeds will depend on our funding requirements and the availability and costs of other funds.
PLAN OF DISTRIBUTION
We may sell the securities from time to time to or through underwriters or dealers, through agents, or directly to one or more purchasers. A distribution of the securities offered by this prospectus may also be effected through the issuance of derivative securities, including without limitation, warrants, rights to purchase and subscriptions. In addition, the manner in which we may sell some or all of the securities covered by this prospectus includes, without limitation, through:
| ● | a block trade in which a broker-dealer will attempt to sell as agent, but may position or resell a portion of the block, as principal, in order to facilitate the transaction; |
| ● | purchases by a broker-dealer, as principal, and resale by the broker-dealer for its account; or |
| ● | ordinary brokerage transactions and transactions in which a broker solicits purchasers. |
A prospectus supplement or supplements with respect to each series of securities will describe the terms of the offering, including, to the extent applicable:
| ● | the terms of the offering; |
| ● | the name or names of the underwriters or agents and the amounts of securities underwritten or purchased by each of them, if any; |
| ● | the public offering price or purchase price of the securities or other consideration therefor, and the proceeds to be received by us from the sale; |
| ● | any delayed delivery requirements; |
| ● | any over-allotment options under which underwriters may purchase additional securities from us; |
| ● | any underwriting discounts or agency fees and other items constituting underwriters’ or agents’ compensation |
| ● | any discounts or concessions allowed or re-allowed or paid to dealers; and |
| ● | any securities exchange or market on which the securities may be listed. |
| 11 |
The offer and sale of the securities described in this prospectus by us, the underwriters or the third parties described above may be effected from time to time in one or more transactions, including privately negotiated transactions, either:
| ● | at a fixed price or prices, which may be changed; |
| ● | in an “at the market” offering within the meaning of Rule 415(a)(4) of the Securities Act of 1933, as amended, or the Securities Act; |
| ● | at prices related to such prevailing market prices; or |
| ● | at negotiated prices. |
Only underwriters named in the prospectus supplement will be underwriters of the securities offered by the prospectus supplement.
Underwriters and Agents; Direct Sales
If underwriters are used in a sale, they will acquire the offered securities for their own account and may resell the offered securities from time to time in one or more transactions, including negotiated transactions, at a fixed public offering price or at varying prices determined at the time of sale. We may offer the securities to the public through underwriting syndicates represented by managing underwriters or by underwriters without a syndicate.
Unless the prospectus supplement states otherwise, the obligations of the underwriters to purchase the securities will be subject to the conditions set forth in the applicable underwriting agreement. Subject to certain conditions, the underwriters will be obligated to purchase all of the securities offered by the prospectus supplement, other than securities covered by any over-allotment option. Any public offering price and any discounts or concessions allowed or re-allowed or paid to dealers may change from time to time. We may use underwriters with whom we have a material relationship. We will describe in the prospectus supplement, naming the underwriter, the nature of any such relationship.
We may sell securities directly or through agents we designate from time to time. We will name any agent involved in the offering and sale of securities, and we will describe any commissions we will pay the agent in the prospectus supplement. Unless the prospectus supplement states otherwise, our agent will act on a best-efforts basis for the period of its appointment.
We may authorize agents or underwriters to solicit offers by certain types of institutional investors to purchase securities from us at the public offering price set forth in the prospectus supplement pursuant to delayed delivery contracts providing for payment and delivery on a specified date in the future. We will describe the conditions to these contracts and the commissions we must pay for solicitation of these contracts in the prospectus supplement.
Dealers
We may sell the offered securities to dealers as principals. The dealer may then resell such securities to the public either at varying prices to be determined by the dealer or at a fixed offering price agreed to with us at the time of resale.
Institutional Purchasers
We may authorize agents, dealers or underwriters to solicit certain institutional investors to purchase offered securities on a delayed delivery basis pursuant to delayed delivery contracts providing for payment and delivery on a specified future date. The applicable prospectus supplement or other offering materials, as the case may be, will provide the details of any such arrangement, including the offering price and commissions payable on the solicitations.
We will enter into such delayed contracts only with institutional purchasers that we approve. These institutions may include commercial and savings banks, insurance companies, pension funds, investment companies and educational and charitable institutions.
Indemnification; Other Relationships
We may provide agents, underwriters, dealers and remarketing firms with indemnification against certain civil liabilities, including liabilities under the Securities Act, or contribution with respect to payments that the agents or underwriters may make with respect to these liabilities. Agents, underwriters, dealers and remarketing firms, and their affiliates, may engage in transactions with, or perform services for, us in the ordinary course of business. This includes commercial banking and investment banking transactions.
| 12 |
Market-Making; Stabilization and Other Transactions
There is currently no market for any of the offered securities, other than our common stock, which is quoted on the Nasdaq Capital Market. If the offered securities are traded after their initial issuance, they may trade at a discount from their initial offering price, depending upon prevailing interest rates, the market for similar securities and other factors. While it is possible that an underwriter could inform us that it intends to make a market in the offered securities, such underwriter would not be obligated to do so, and any such market-making could be discontinued at any time without notice. Therefore, no assurance can be given as to whether an active trading market will develop for the offered securities. We have no current plans for listing of the preferred stock, warrants or subscription rights on any securities exchange or quotation system; any such listing with respect to any particular preferred stock, warrants or subscription rights will be described in the applicable prospectus supplement or other offering materials, as the case may be.
Any underwriter may engage in over-allotment, stabilizing transactions, short-covering transactions and penalty bids in accordance with Regulation M under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Over-allotment involves sales in excess of the offering size, which create a short position. Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum price. Syndicate-covering or other short-covering transactions involve purchases of the securities, either through exercise of the over-allotment option or in the open market after the distribution is completed, to cover short positions. Penalty bids permit the underwriters to reclaim a selling concession from a dealer when the securities originally sold by the dealer are purchased in a stabilizing or covering transaction to cover short positions. Those activities may cause the price of the securities to be higher than it would otherwise be. If commenced, the underwriters may discontinue any of the activities at any time.
Any underwriters or agents that are qualified market makers on the Nasdaq Capital Market may engage in passive market making transactions in our common stock on the Nasdaq Capital Market in accordance with Regulation M under the Exchange Act, during the business day prior to the pricing of the offering, before the commencement of offers or sales of our common stock. Passive market makers must comply with applicable volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Fees and Commissions
If 5% or more of the net proceeds of any offering of securities made under this prospectus will be received by a FINRA member participating in the offering or affiliates or associated persons of such FINRA member, the offering will be conducted in accordance with FINRA Rule 5121.
DESCRIPTION OF SECURITIES WE MAY OFFER
The descriptions of the securities contained in this prospectus, together with the applicable prospectus supplements, summarize all the material terms and provisions of the various types of securities that we may offer. We will describe in the applicable prospectus supplement relating to any securities the particular terms of the securities offered by that prospectus supplement. If we indicate in the applicable prospectus supplement, the terms of the securities may differ from the terms we have summarized below. We will also include in the prospectus supplement information, where applicable, about material United States federal income tax considerations relating to the securities, and the securities exchange, if any, on which the securities will be listed.
We may sell from time to time, in one or more offerings:
| • | shares of our common stock; |
| • | shares of our preferred stock; |
| • | warrants to purchase shares of our common stock or preferred stock; |
| • | rights to purchase shares of our common stock; and/or |
| • | units consisting of any of the securities listed above. |
The terms of any securities we offer will be determined at the time of sale. We may issue securities that are exchangeable for or convertible into common stock or any of the other securities that may be sold under this prospectus. When particular securities are offered, a supplement to this prospectus will be filed with the Commission, which will describe the terms of the offering and sale of the offered securities.
| 13 |
DESCRIPTION OF CAPITAL STOCK
The summary does not purport to be complete and is qualified in its entirety by reference to our certificate of incorporation and bylaws, and to the provisions of the General Corporation Law of the State of Delaware, as amended.
We are authorized to issue 300,000,000 shares of common stock, par value $0.0001 per share. As of the date of this prospectus, there were 96,427,624 shares of our common stock issued and outstanding. The outstanding shares of our common stock are validly issued, fully paid and nonassessable. We are authorized to issue up to 10,000,000 shares of preferred stock, par value $0.0001 per share. Of these shares of preferred stock, 1,360,000 are designated as Series A Convertible Preferred Stock. As of the date of this prospectus, there were no shares of Series A Convertible Preferred Stock issued or outstanding.
Common Stock
Holders of our shares of common stock are entitled to one vote for each share on all matters submitted to a shareholder vote. Holders of our common stock do not have cumulative voting rights. Therefore, holders of a majority of the shares of our common stock voting for the election of directors can elect all of the directors. Holders of our common stock representing a majority of the voting power of our capital stock issued, outstanding and entitled to vote, represented in person or by proxy, are necessary to constitute a quorum at any meeting of shareholders. A vote by the holders of a majority of our outstanding shares is required to effectuate certain fundamental corporate changes such as liquidation, merger or an amendment to our certificate of incorporation.
Holders of our common stock are entitled to share in all dividends that our board of directors, in its discretion, declares from legally available funds. In the event of a liquidation, dissolution or winding up, each outstanding share entitles its holder to participate pro rata in all assets that remain after payment of liabilities and after providing for each class of stock, if any, having preference over our common stock. Our common stock has no preemptive, subscription or conversion rights and there are no redemption provisions applicable to our common stock.
Preferred Stock
The shares of preferred stock may be issued in series, and shall have such voting powers, full or limited, or no voting powers, and such designations, preferences and relative participating, optional or other special rights, and qualifications, limitations or restrictions thereof, as shall be stated and expressed in the resolution or resolutions providing for the issuance of such stock adopted from time to time by the board of directors. The board of directors is expressly vested with the authority to determine and fix in the resolution or resolutions providing for the issuances of preferred stock the voting powers, designations, preferences and rights, and the qualifications, limitations or restrictions thereof, of each such series to the full extent now or hereafter permitted by the laws of the State of Delaware.
The authorized shares of preferred stock will be available for issuance without further action by our stockholders unless such action is required by applicable law or the rules of any stock exchange or automated quotation system on which our securities may be listed or traded. The Nasdaq Stock Market currently requires stockholder approval as a prerequisite to listing shares in several circumstances, including, in certain circumstances, where the issuance of shares could result in an increase in the number of shares of common stock outstanding, or in the amount of voting securities outstanding, of at least 20%.
Transfer Agent and Registrar
The Transfer Agent and Registrar for our common stock is Computershare Trust Company, N.A., 8742 Lucent Blvd., Suite 225, Highlands Ranch, CO 80129.
| 14 |
DESCRIPTION OF WARRANTS
The following description, together with the additional information we may include in any applicable prospectus supplements, summarizes the material terms and provisions of the warrants that we may offer under this prospectus and the related warrant agreements and warrant certificates. While the terms summarized below will apply generally to any warrants that we may offer, we will describe the particular terms of any series of warrants in more detail in the applicable prospectus supplement. If we indicate in the prospectus supplement, the terms of any warrants offered under that prospectus supplement may differ from the terms described below. If there are differences between that prospectus supplement and this prospectus, the prospectus supplement will control. Thus, the statements we make in this section may not apply to a particular series of warrants. Specific warrant agreements will contain additional important terms and provisions and will be incorporated by reference as an exhibit to the registration statement which includes this prospectus.
General
We may issue warrants for the purchase of common stock and/or preferred stock in one or more series. We may issue warrants independently or together with common stock and/or preferred stock, and the warrants may be attached to or separate from these securities.
We will evidence each series of warrants by warrant certificates that we may issue under a separate agreement. We may enter into the warrant agreement with a warrant agent. Each warrant agent may be a bank that we select which has its principal office in the United States and a combined capital and surplus of at least $50,000,000. We may also choose to act as our own warrant agent. We will indicate the name and address of any such warrant agent in the applicable prospectus supplement relating to a particular series of warrants.
We will describe in the applicable prospectus supplement the terms of the series of warrants, including:
| • | the offering price and aggregate number of warrants offered; |
| • | the currency for which the warrants may be purchased; |
| • | if applicable, the designation and terms of the securities with which the warrants are issued and the number of warrants issued with each such security or each principal amount of such security; |
| • | if applicable, the date on and after which the warrants and the related securities will be separately transferable; |
| • | in the case of warrants to purchase common stock or preferred stock, the number of shares of common stock or preferred stock, as the case may be, purchasable upon the exercise of one warrant and the price at which these shares may be purchased upon such exercise; |
| • | the warrant agreement under which the warrants will be issued; |
| • | the effect of any merger, consolidation, sale or other disposition of our business on the warrant agreement and the warrants; |
| • | anti-dilution provisions of the warrants, if any; |
| • | the terms of any rights to redeem or call the warrants; |
| • | any provisions for changes to or adjustments in the exercise price or number of securities issuable upon exercise of the warrants; |
| • | the dates on which the right to exercise the warrants will commence and expire or, if the warrants are not continuously exercisable during that period, the specific date or dates on which the warrants will be exercisable; |
| • | the manner in which the warrant agreement and warrants may be modified; |
| • | the identities of the warrant agent and any calculation or other agent for the warrants; |
| • | federal income tax consequences of holding or exercising the warrants; |
| • | the terms of the securities issuable upon exercise of the warrants; |
| 15 |
| • | any securities exchange or quotation system on which the warrants or any securities deliverable upon exercise of the warrants may be listed; and |
| • | any other specific terms, preferences, rights or limitations of or restrictions on the warrants. |
Before exercising their warrants, holders of warrants will not have any of the rights of holders of the securities purchasable upon such exercise, including in the case of warrants to purchase common stock or preferred stock, the right to receive dividends, if any, or, payments upon our liquidation, dissolution or winding up or to exercise voting rights, if any.
Exercise of Warrants
Each warrant will entitle the holder to purchase the securities that we specify in the applicable prospectus supplement at the exercise price that we describe in the applicable prospectus supplement. Unless we otherwise specify in the applicable prospectus supplement, holders of the warrants may exercise the warrants at any time up to 5:00 p.m. Eastern Time on the expiration date that we set forth in the applicable prospectus supplement. After the close of business on the expiration date, unexercised warrants will become void.
Holders of the warrants may exercise the warrants by delivering the warrant certificate representing the warrants to be exercised together with specified information, and paying the required amount to the warrant agent in immediately available funds, as provided in the applicable prospectus supplement. We will set forth on the reverse side of the warrant certificate, and in the applicable prospectus supplement, the information that the holder of the warrant will be required to deliver to the warrant agent.
Until the warrant is properly exercised, no holder of any warrant will be entitled to any rights of a holder of the securities purchasable upon exercise of the warrant.
Upon receipt of the required payment and the warrant certificate properly completed and duly executed at the corporate trust office of the warrant agent or any other office indicated in the applicable prospectus supplement, we will issue and deliver the securities purchasable upon such exercise. If fewer than all of the warrants represented by the warrant certificate are exercised, then we will issue a new warrant certificate for the remaining amount of warrants. If we so indicate in the applicable prospectus supplement, holders of the warrants may surrender securities as all or part of the exercise price for warrants.
Enforceability of Rights by Holders of Warrants
Any warrant agent will act solely as our agent under the applicable warrant agreement and will not assume any obligation or relationship of agency or trust with any holder of any warrant. A single bank or trust company may act as warrant agent for more than one issue of warrants. A warrant agent will have no duty or responsibility in case of any default by us under the applicable warrant agreement or warrant, including any duty or responsibility to initiate any proceedings at law or otherwise, or to make any demand upon us. Any holder of a warrant may, without the consent of the related warrant agent or the holder of any other warrant, enforce by appropriate legal action its right to exercise, and receive the securities purchasable upon exercise of, its warrants in accordance with their terms.
Warrant Agreement Will Not Be Qualified Under the Trust Indenture Act
No warrant agreement will be qualified as an indenture, and no warrant agent will be required to qualify as a trustee, under the Trust Indenture Act. Therefore, holders of warrants issued under a warrant agreement will not have the protection of the Trust Indenture Act with respect to their warrants.
Governing Law
Each warrant agreement and any warrants issued under the warrant agreements will be governed by New York law.
Calculation Agent
Calculations relating to warrants may be made by a calculation agent, an institution that we appoint as our agent for this purpose. The prospectus supplement for a particular warrant will name the institution that we have appointed to act as the calculation agent for that warrant as of the original issue date for that warrant. We may appoint a different institution to serve as calculation agent from time to time after the original issue date without the consent or notification of the holders.
The calculation agent’s determination of any amount of money payable or securities deliverable with respect to a warrant will be final and binding in the absence of manifest error.
| 16 |
DESCRIPTION OF RIGHTS
This section describes the general terms of the rights that we may offer and sell by this prospectus. This prospectus and any accompanying prospectus supplement will contain the material terms and conditions for each right. The accompanying prospectus supplement may add, update or change the terms and conditions of the rights as described in this prospectus.
The particular terms of each issue of rights, the rights agreement relating to the rights and the rights certificates representing rights will be described in the applicable prospectus supplement, including, as applicable:
| • | the title of the rights; |
| • | the date of determining the stockholders entitled to the rights distribution; |
| • | the title, aggregate number of shares of common stock or preferred stock purchasable upon exercise of the rights; |
| • | the exercise price; |
| • | the aggregate number of rights issued; |
| • | the date, if any, on and after which the rights will be separately transferable; |
| • | the date on which the right to exercise the rights will commence and the date on which the right will expire; and |
| • | any other terms of the rights, including terms, procedures and limitations relating to the distribution, exchange and exercise of the rights. |
DESCRIPTION OF UNITS
We may issue units comprised of one or more of the other securities described in this prospectus in any combination. Each unit will be issued so that the holder of the unit is also the holder of each security included in the unit. Thus, the holder of a unit will have the rights and obligations of a holder of each included security. The unit agreement under which a unit is issued may provide that the securities included in the unit may not be held or transferred separately, at any time or at any time before a specified date.
The applicable prospectus supplement will describe:
| • | the designation and terms of the units and of the securities comprising the units, including whether and under what circumstances those securities may be held or transferred separately; |
| • | any unit agreement under which the units will be issued; |
| • | any provisions for the issuance, payment, settlement, transfer or exchange of the units or of the securities comprising the units; and |
| • | whether the units will be issued in fully registered or global form. |
The applicable prospectus supplement will describe the terms of any units. The preceding description and any description of units in the applicable prospectus supplement does not purport to be complete and is subject to and is qualified in its entirety by reference to the unit agreement and, if applicable, collateral arrangements and depositary arrangements relating to such units.
| 17 |
LEGAL MATTERS
The validity of the securities offered by this prospectus is being passed upon for us by our counsel, Olshan Frome Wolosky LLP, New York, New York. If the securities are distributed in an underwritten offering, certain legal matters will be passed upon for the underwriters by counsel identified in the applicable prospectus supplement.
EXPERTS
The financial statements of Alzamend Neuro, Inc. as of April 30, 2023 and 2022 and for each of the two years in the period ended April 30, 2023 incorporated by reference in this Prospectus and Registration Statement from our Annual Report on Form 10-K for the years ended April 30, 2023 and 2022, have been audited by Baker Tilly US, LLP, an independent registered public accounting firm, as stated in their report thereon (which report expresses an unqualified opinion and includes an explanatory paragraph relating to the Company’s ability to continue as a going concern), incorporated herein by reference, and have been incorporated in this Prospectus and Registration Statement in reliance upon such report and upon the authority of such firm as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the Commission a registration statement on Form S-3 under the Securities Act, with respect to the securities covered by this prospectus. This prospectus and any prospectus supplement which form a part of the registration statement, does not contain all of the information set forth in the registration statement or the exhibits and schedules filed therewith. For further information with respect to us and the securities covered by this prospectus, please see the registration statement and the exhibits filed with the registration statement. Any statements made in this prospectus or any prospectus supplement concerning legal documents are not necessarily complete and you should read the documents that are filed as exhibits to the registration statement or otherwise filed with the Commission for a more complete understanding of the document or matter. A copy of the registration statement and the exhibits filed with the registration statement may be inspected without charge at the Public Reference Room maintained by the Commission, located at 100 F Street, N.E., Washington, D.C. 20549. Please call the Commission at 1-800-SEC-0330 for more information about the operation of the Public Reference Room. The Commission also maintains an internet website that contains reports, proxy and information statements and other information regarding registrants that file electronically with the Commission. The address of the website is http://www.sec.gov.
We file annual, quarterly and current reports, proxy statements and other information with the Commission. You may read, without charge, and copy the documents we file at the Commission’s public reference room in Washington, D.C. at 100 F Street, N.E., Washington, D.C. 20549. You can request copies of these documents by writing to the Commission and paying a fee for the copying cost. Please call the Commission at 1-800-SEC-0330 for further information on the public reference rooms. Our filings with the Commission are available to the public at no cost from the Commission’s website at http://www.sec.gov.
| 18 |
INCORPORATION OF DOCUMENTS BY REFERENCE
We are “incorporating by reference” in this prospectus certain documents we file with the Commission, which means that we can disclose important information to you by referring you to those documents. The information in the documents incorporated by reference is considered to be part of this prospectus. Statements contained in documents that we file with the Commission and that are incorporated by reference in this prospectus will automatically update and supersede information contained in this prospectus, including information in previously filed documents or reports that have been incorporated by reference in this prospectus, to the extent the new information differs from or is inconsistent with the old information. We have filed the following document with the Commission, which is incorporated herein by reference as of its date of filing:
| • | Our Annual Report on Form 10-K for the period ended April 30, 2023, filed with the Commission on July 27, 2023. |
All documents that we filed with the Commission pursuant to Sections 13(a), 13(c), 14, and 15(d) of the Exchange Act subsequent to the date of this registration statement and prior to the filing of a post-effective amendment to this registration statement that indicates that all securities offered under this prospectus have been sold, or that deregisters all securities then remaining unsold, will be deemed to be incorporated in this registration statement by reference and to be a part hereof from the date of filing of such documents.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus shall be deemed modified, superseded or replaced for purposes of this prospectus to the extent that a statement contained in this prospectus, or in any subsequently filed document that also is deemed to be incorporated by reference in this prospectus, modifies, supersedes or replaces such statement. Any statement so modified, superseded or replaced shall not be deemed, except as so modified, superseded or replaced, to constitute a part of this prospectus. None of the information that we disclose under Items 2.02 or 7.01 of any Current Report on Form 8-K or any corresponding information, either furnished under Item 9.01 or included as an exhibit therein, that we may from time to time furnish to the Commission will be incorporated by reference into, or otherwise included in, this prospectus, except as otherwise expressly set forth in the relevant document. Subject to the foregoing, all information appearing in this prospectus is qualified in its entirety by the information appearing in the documents incorporated by reference.
You may request, orally or in writing, a copy of these documents, which will be provided to you at no cost (other than exhibits, unless such exhibits are specifically incorporate by reference), by contacting Stephan Jackman, c/o Alzamend Neuro, Inc., at 3480 Peachtree Road NE, Second Floor, Suite 103, Atlanta, GA 30326. Our telephone number is (844) 722-6333. Information about us is also available at our website at www.alzamend.com. However, the information on our website is not a part of this prospectus and is not incorporated by reference.
| 19 |
Up to $9,831,947
Alzamend Neuro, Inc.
Shares of Common Stock
PROSPECTUS SUPPLEMENT
![]()
____________________
The date of this Prospectus Supplement is September 8, 2023